kr
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10‑K
|
|
|
|
(Mark One)
|
|
|
☒
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
For the fiscal year ended December 31, 2015
|
|
or
|
|
☐
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
Commission file number 0‑29889
RIGEL PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
|
|
|
|
Delaware
(State or other jurisdiction of
incorporation or organization)
|
94‑3248524
(IRS Employer
Identification No.)
|
|
1180 Veterans Blvd.
South San Francisco, California
(Address of principal executive offices)
|
94080
(Zip Code)
|
(650) 624‑1100
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
Title of each class:
|
Name of each exchange on which registered:
|
|
Common Stock, par value $.001 per share
|
The Nasdaq Global Market
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well‑known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S‑T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S‑K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10‑K or any amendment to this Form 10‑K. ☒
Indicate by a check mark whether the registrant is a large accelerated filer, an accelerated filer, a non‑accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b‑2 of the Exchange Act).
|
|
|
|
|
|
Large accelerated filer ☐
|
Accelerated filer ☒
|
Non‑accelerated filer ☐
(Do not check if a
smaller reporting company)
|
Smaller reporting company ☐
|
Indicate by a check mark whether the registrant is a shell company (as defined in Rule 12b‑2 of the Act). Yes ☐ No ☒
The approximate aggregate market value of the Common Stock held by non‑ affiliates of the registrant, based upon the closing price of the registrant’s Common Stock as reported on the Nasdaq Global Market on June 30, 2015, the last business day of the registrant’s most recently completed second fiscal quarter, was $283,029,401. Shares of the registrant’s outstanding Common Stock held by each executive officer, director and affiliates of the registrant’s outstanding Common Stock have been excluded. The determination of affiliate status for the purposes of this calculation is not necessarily a conclusive determination for other purposes.
As of February 29, 2016, there were 90,556,255 shares of the registrant’s Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Items 10, 11, 12, 13 and 14 of Part III of this Annual Report on Form 10‑K incorporate information by reference from the definitive proxy statement for the registrant’s 2016 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10‑K.
FORWARD‑LOOKING STATEMENTS
This Annual Report on Form 10‑K contains statements indicating expectations about future performance and other forward‑looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the Securities Act), Section 21E of the Securities Exchange Act of 1934, as amended (the Exchange Act), and the Private Securities Litigation Reform Act of 1995, that involve risks and uncertainties. We usually use words such as “may,” “will,” “should,” “could,” “expect,” “plan,” “anticipate,” “might,” “believe,” “estimate,” “predict,” “intend” or the negative of these terms or similar expressions to identify these forward‑ looking statements. These statements appear throughout this Annual Report on Form 10‑K and are statements regarding our current intent, belief or expectation, primarily with respect to our operations and related industry developments. Examples of these statements include, but are not limited to, statements regarding the following: our business and scientific strategies; the progress of our product development programs, including clinical testing, and the timing of commencement and results thereof; our corporate collaborations, and revenues that may be received from collaborations and the timing of those potential payments; our drug discovery technologies; our research and development expenses; protection of our intellectual property; and sufficiency of our cash resources and need for additional capital. You should not place undue reliance on these forward‑looking statements. Our actual results could differ materially from those anticipated in these forward‑looking statements for many reasons, including as a result of the risks and uncertainties discussed under the heading “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10‑K. A forward‑ looking statement speaks only as of the date on which it is made, and, except as required by law, we undertake no obligation to update any forward‑looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events. New factors emerge from time to time, and it is not possible for us to predict which factors will arise. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward‑looking statements.
PART I
Item 1. Business
Overview
Rigel Pharmaceuticals, Inc. was incorporated in Delaware in June 1996, and is based in South San Francisco, California. We are a clinical‑stage biotechnology company dedicated to the discovery and development of novel, targeted drugs in the therapeutic areas of immunology, oncology and immuno-oncology. Our pioneering research focuses on signaling pathways that are critical to disease mechanisms. Our current clinical programs include fostamatinib, an oral spleen tyrosine kinase (SYK) inhibitor, which is in Phase 3 clinical trials for immune thrombocytopenic purpura (ITP); a Phase 2 clinical trial for autoimmune hemolytic anemia (AIHA); and a Phase 2 clinical trial for IgA nephropathy (IgAN). In addition, we have two oncology product candidates in Phase 1 development with partners BerGenBio AS (BergenBio) and Daiichi Sankyo (Daiichi).
Since the beginning of 2015, we have experienced the following significant business events:
|
·
| |
In February 2016, we announced that we initiated a Phase 2 clinical trial to evaluate fostamatinib as a potential treatment for AIHA. The trial is a two-stage study and we expect to report the results of the Stage 1 segment by the end of 2016. |
|
·
| |
In January 2016, we experienced the following fostamatinib events: |
|
i.)
| |
we completed patient enrollment of the first of two Phase 3 studies with fostamatinib for the treatment of ITP and expect to report results from the first study in the middle of 2016. We expect to report results on the second study shortly thereafter; |
|
ii.)
| |
we announced that the Phase 2 study of fostamatinib in IgAN continues to enroll patients for the first cohort in various centers throughout Asia, the U.S. and Europe, and that the study is on track to report top line results in the second half of 2016; and |
|
·
| |
In September 2015, we announced that we entered into an exclusive, worldwide license agreement with Aclaris Therapeutics International Limited (Aclaris) for the development and commercialization of certain JAK inhibitors for the treatment of alopecia areata and other dermatological conditions. |
|
·
| |
In September 2015, we announced that the U.S. Food and Drug Administration (FDA) has granted Orphan Drug designation to fostamatinib for the treatment of ITP. |
|
·
| |
In June 2015, we announced that Keith A. Katkin, president and chief executive officer of Avanir Pharmaceuticals, has been appointed to our board of directors. |
|
·
| |
In February 2015, we announced that we entered into a collaboration agreement with Bristol-Myers Squibb Company (BMS) for the discovery, development and commercialization of cancer immunotherapies based on our extensive portfolio of small molecule TGF beta receptor kinase inhibitors, in which BMS paid us an upfront payment of $30.0 million. |
|
·
| |
In January 2015, we announced that in December 2014 we earned a non‑refundable payment of $5.8 million from AZ resulting from AZ’s continued development of R256 in asthma as of December 2014, which we received in the first quarter of 2015. |
Strategy
Our research team is focused on creating a portfolio of product candidates that may be developed as therapeutics for our own proprietary programs or for development by potential collaborative partners. We recognize that the product development process is subject to both high costs and a high risk of failure. We believe that identifying a variety of product candidates and working in conjunction with other pharmaceutical partners may minimize the risk of failure, fill the product pipeline gap at major pharmaceutical companies, and ultimately increase the likelihood of advancing clinical development and potential commercialization of the product candidates.
The key elements to our business and scientific strategy are to:
|
·
| |
develop and commercialize fostamatinib in the United States where we believe a company our size can successfully compete; |
|
·
| |
outlicense European and Asian rights to fostamatinib with Phase 3 clinical data in hand; |
|
·
| |
develop and commercialize fostamatinib for possible additional indications; |
|
·
| |
develop a diverse portfolio of drug candidates that address a focused band of therapeutic indications or that represent significant market opportunities; |
|
·
| |
utilize our robust discovery engine to rapidly discover and validate new product candidates in a focused range of therapeutic indications; and |
|
·
| |
develop drug candidates and establish strategic collaborations with pharmaceutical and biotechnology companies to further develop and market our product candidates. |
Product Development Programs
Our product development portfolio features multiple novel, targeted drug candidates in the therapeutic areas of immunology, oncology and immuno-oncology.
|
|
|
|
|
|
|
|
Pipeline
|
|
Current Stage
|
|
Status
|
|
Fostamatinib—Oral SYK Inhibitor
|
|
|
|
|
|
Immune Thrombocytopenic Purpura (ITP)
|
|
Phase 3
|
|
We completed patient enrollment of the first of two Phase 3 studies with fostamatinib for the treatment of ITP in January 2016 and expect to report results from the first study in the middle of 2016. We expect to report results on the second study shortly thereafter.
|
|
IgA Nephropathy (IgAN)
|
|
Phase 2
|
|
The Phase 2 study of fostamatinib in IgAN continues to enroll patients for the first cohort in various centers throughout Asia, the U.S. and Europe, and that the study is on track to report top line results in the second half of 2016.
|
|
Autoimmune Hemolytic Anemia (AIHA)
|
|
Phase 2
|
|
We initiated a Phase 2 clinical trial in patients with AIHA in February 2016. The trial is a two-stage study and we expect to report the results of the Stage 1 segment by the end of 2016.
|
|
R348—Topical Ophthalmic JAK/SYK Inhibitor
|
|
|
|
|
|
Dry Eye in Patients with Ocular Graft‑Versus‑Host Disease (GvHD)
|
|
Phase 2
|
|
R348 is being evaluated in a Phase 2 clinical trial of patients with ocular GvHD to determine if it reduces inflammation and limits the damage to the eye tissue caused by the disease. We expect results of this clinical trial in 2016.
|
Clinical Stage Programs
Fostamatinib—Immune Thrombocytopenic Purpura
Disease background. Chronic ITP affects an estimated 60,000 to 125,000 people in the U.S. In patients with ITP, the immune system attacks and destroys the body’s own blood platelets, which play an active role in blood clotting and healing. ITP patients can suffer extraordinary bruising, bleeding and fatigue as a result of low platelet counts.
Current therapies for ITP include steroids, blood platelet production boosters that imitate thrombopoietin (TPOs) and splenectomy.
Orally‑available SYK inhibitor program. Taken in tablet form, fostamatinib blocks the activation of SYK inside immune cells. ITP causes the body to produce antibodies that attach to healthy platelets in the blood stream. Immune cells recognize these antibodies and affix to them, which activates the SYK enzyme inside the immune cell, and triggers the destruction of the antibody and the attached platelet. When SYK is inhibited by fostamatinib, it interrupts this immune cell function and allows the platelets to escape destruction. The results of our Phase 2 clinical trial, in which fostamatinib was orally administered to sixteen adults with chronic ITP, published in Blood, showed that fostamatinib significantly increased the platelet counts of certain ITP patients, including those who had failed other currently available agents.
In October 2013, we met with the U.S. FDA for an end-of-Phase 2 meeting for fostamatinib in ITP. Based on that meeting, we designed a Phase 3 clinical program, called fostamatinib in thrombocytopenia (FIT), in which a total of 150 ITP patients will be randomized into two identical multi-center, double-blind, placebo-controlled clinical trials. The patients will have been diagnosed with persistent or chronic ITP, and have blood platelet counts consistently below 30,000 per microliter of blood. Two-thirds of the subjects will receive fostamatinib orally at 100 mg bid (twice daily) and the other third will receive placebo on the same schedule. Subjects are expected to remain on treatment for 24 weeks. At week four of treatment, subjects who meet certain platelet count and tolerability thresholds will have their dosage of fostamatinib (or corresponding placebo) increased to 150 mg bid. The primary efficacy endpoint of this program is a stable platelet response by week 24 with platelet counts at or above 50,000 per microliter of blood for at least four of the final six qualifying blood draws. In August 2015, the FDA granted our request for Orphan Drug designation to fostamatinib, our oral SYK inhibitor, for the treatment of ITP. The first trial of our Phase 3 clinical program for ITP completed patient enrollment in January 2016. Our second Phase 3 trial is currently actively enrolling patients. We expect to separately report top line results of the two Phase 3 trials, with the first trial reporting in the middle of 2016 and the other trial reporting shortly thereafter.
Fostamatinib—IgAN
Disease background. IgAN is an autoimmune disease that severely affects the functioning of the kidneys. An estimated 12,000 Americans are diagnosed with this type of glomerulonephritis each year, with 25% of its victims eventually requiring dialysis and/or kidney transplantation over time. IgAN is characterized by the deposition of IgA immune complexes in the glomeruli of the kidneys leading to an inflammatory response and subsequent tissue damage that ultimately disrupts the normal filtering function of the kidneys. By inhibiting SYK in kidney cells, fostamatinib may block the signaling of IgA immune complex receptors and arrest or slow destruction of the glomeruli.
Orally-available SYK inhibitor program. Our Phase 2 clinical trial in patients with IgAN, called SIGN (SYK Inhibition for Glomerulonephritis) continues to enroll patients for the first cohort. We expect to report top line results in the second half of 2016.
Fostamatinib—AIHA
Disease background.
AIHA is a rare, serious blood disorder where the immune system produces antibodies that result in the destruction of the body's own red blood cells. Symptoms can include fatigue, shortness of breath, rapid heartbeat, jaundice or enlarged spleen. While no medical treatments are currently approved for AIHA, physicians generally treat acute and chronic cases of the disorder with corticosteroids, other immuno-suppressants, or splenectomy. Research has shown that inhibiting SYK with fostamatinib may reduce the destruction of red blood cells. This disorder affects an estimated 40,000 Americans, for whom no approved treatment options currently exist.
Orally‑available SYK inhibitor program. We initiated a Phase 2 clinical trial in patients with AIHA in February 2016. The trial is an open-label, multi-center, two-stage study that will evaluate the efficacy and safety of fostamatinib in patients with warm antibody AIHA who have previously received treatment for the disorder, but have relapsed. Stage 1
will enroll 17 patients who will receive 150 mg of fostamatinib orally twice a day for a period of 12 weeks. The patients will return to the clinic every two weeks for blood draws and medical assessment. The primary efficacy endpoint of this study is to achieve increased hemoglobin levels by week 12 of greater than 10 g/dL, and greater than or equal to 2 g/dL higher than baseline. Stage 2 will begin after enrollment in Stage 1 has been completed and will include an additional 20 patients who will receive the same treatment protocol as Stage 1. We expect to have results of the Stage 1 segment of the trial by the end of 2016.
R348—Dry Eye in Patients with Ocular Graft‑Versus‑Host Disease (GvHD)
Disease background. According to an article published by the American Academy of Ophthalmology, a significant number (22% to 80%) of patients with acute or chronic GvHD develop a secondary incidence of dry eye (keratoconjunctivitis sicca). In general, these patients are severely ill and have a great medical need for a topical therapy that may better manage their symptoms.
Topical Ophthalmic JAK/SYK inhibitor program. R348, an ophthalmic JAK/SYK inhibitor, is being evaluated in a Phase 2 study of patients with ocular GvHD to determine if it reduces inflammation and limits the damage to the eye tissue caused by the disease. We expect results of this clinical trial in 2016.
Research/Preclinical Programs
We are conducting proprietary research in the broad disease areas of inflammation/immunology, immuno-oncology, cancers and muscle wasting/muscle endurance. Within each disease area, our researchers are investigating mechanisms of action as well as screening compounds against potential novel targets and optimizing those leads that appear to have the greatest potential.
We are conducting preclinical studies to identify a lead molecule for our IRAK program. This program may provide opportunities in both the oncology and immunology areas, including acute myeloid leukemia (AML). We are currently targeting AML and MDS with different mechanisms of action in various preclinical projects.
Leveraging our extensive immunology expertise, we are continuing to explore novel immuno-oncology approaches to treating various oncology indications. The first of these resulted in a collaboration with BMS for TGF beta receptor kinase inhibitors. Several other projects are currently underway.
Sponsored Research and License Agreements
We conduct research and development programs independently and in connection with our corporate collaborators. We are a participant in our collaboration agreement with BMS for the discovery, development and commercialization of cancer immunotherapies based on our small molecule TGF beta receptor kinase inhibitors, as discussed below. Our participation is limited to the Joint Research Committee and the performance of research activities based on billable full-time equivalent fees as specified in the agreement. We do not have ongoing participation obligations under our agreements with Aclaris for the development and commercialization of certain JAK inhibitors for the treatment of alopecia areata and other dermatological conditions, AZ for the development and commercialization of R256, an inhaled JAK inhibitor, BerGenBio for the development and commercialization of an oncology program, and Daiichi to pursue research related to a specific target from a novel class of drug targets called ligases. Under these agreements, which we entered into in the ordinary course of business, we received or may be entitled to receive upfront cash payments, progress dependent contingent payments on events achieved by such partners and royalties on any net sales of products sold by such partners under the agreements. Total future contingent payments to us under all of these current agreements could exceed $533.6 million if all potential product candidates achieved all of the payment triggering events under all of our current agreements (based on a single product candidate under each agreement). Of this amount, up to $150.5 million relates to the achievement of development events, up to $345.6 million relates to the achievement of regulatory events and up to $37.5 million relates to the achievement of certain commercial or launch events. This estimated future contingent amount does not include any estimated royalties that could be due to us if the partners successfully commercialize any of the licensed products. Future events that may trigger payments to us under the
agreements are based solely on our partners’ future efforts and achievements of specified development, regulatory and/or commercial events.
Because we do not control the research, development or commercialization of the product candidates generated under these agreements, we are not able to reasonably estimate when, if at all, any contingent payments would become payable to us. As such, the contingent payments we could receive thereunder involve a substantial degree of risk to achieve and may never be received in the next twelve months or thereafter. Accordingly, we do not expect, and investors should not assume, that we will receive all of the potential contingent payments provided for under these agreements and it is possible that we may never receive any additional significant contingent payments or royalties under these agreements.
In October 2015, we entered into a non-exclusive license agreement with a third party, pursuant to which we received a payment in the single-digit millions in exchange for granting a non-exclusive license to certain limited intellectual property rights. We concluded that the granting of the license, which was fully delivered to such third party in the fourth quarter of 2015, represents the sole deliverable under this agreement. Accordingly, we have recognized the payment as revenue during the year ended December 31, 2015.
In August 2015, we entered into a license agreement with Aclaris, pursuant to which Aclaris will have exclusive rights and will assume responsibility for the continued development of certain JAK inhibitor compounds for the treatment of alopecia areata and other dermatological conditions. Under the license agreement, we received a noncreditable and non-refundable upfront payment of $8.0 million in September 2015. We are also entitled to receive development and regulatory contingent fees that could exceed $80.0 million for a successful compound approved in certain indications. In addition, we are also eligible to receive tiered royalties on the net sales of any products under the agreement. We concluded that the granting of the license, which has been fully delivered to Aclaris in the third quarter of 2015, represents the sole deliverable under this agreement. Accordingly, we have recognized the $8.0 million payment as revenue during the year ended December 31, 2015.
In February 2015, we entered into a collaboration agreement with BMS for the discovery, development and commercialization of cancer immunotherapies based on our extensive portfolio of small molecule TGF beta receptor kinase inhibitors. Under the collaboration agreement, BMS will have exclusive rights and will be solely responsible for the clinical development and commercialization of any products. Pursuant to the collaboration agreement with BMS, we received a noncreditable and non-refundable upfront payment of $30.0 million in March 2015. We are also entitled to receive development and regulatory contingent fees that could exceed $309.0 million for a successful compound approved in certain indications. In addition, we are also eligible to receive tiered royalties on the net sales of any products from the collaboration. BMS shall also reimburse us for agreed upon costs based on a contractual cost per full-time equivalent employee in connection with the performance of research activities during the research term. Under the collaboration agreement, we were obligated to provide the following deliverables: (i) granting of license rights to our program, (ii) participation in the Joint Research Committee, and (iii) performance of research activities. We concluded that these deliverables are a single unit of accounting as the license does not have stand-alone value apart from the other deliverables. Accordingly, the $30.0 million upfront payment is being recognized ratably as revenue from the effective date of the agreement through September 2016, the end of the estimated research term. We believe that straight-line recognition of this revenue is appropriate as the research is expected to be performed ratably over the research period. During the year ended December 31, 2015, we recognized revenue of $16.6 million and $822,000 relating to the upfront payment and research activities we performed, respectively. As of December 31, 2015, deferred revenue related to the $30.0 million upfront payment was $13.4 million.
Our Discovery Engine
The approaches that we use in connection with both our proprietary product development programs and our corporate collaborations are designed to identify protein targets for compound screening and validate the role of those targets in the disease process. Unlike genomics‑based approaches, which begin by identifying genes and then searching for their functions, our approach identifies proteins that are demonstrated to have an important role in a specific disease pathway. By understanding the disease pathway, we attempt to avoid studying genes that will not make good drug
targets and focus only on the subset of expressed proteins of genes that we believe are specifically implicated in the disease process.
We begin by developing assays that model the key events in a disease process at the cellular level. We then identify potential protein targets. In addition, we identify the proteins involved in the intracellular process and prepare a map of their interactions, thus giving us a comprehensive picture of the intracellular disease pathway. We believe that our approach has a number of advantages, including:
|
·
| |
improved target identification: it focuses only on the subset of expressed proteins of genes believed to be specifically implicated in the disease process; |
|
·
| |
rapid validation of protein targets: it produces validated protein targets quickly because it uses key events in the disease process as the basis to design the functional, disease‑based screen; |
|
·
| |
improved disease pathway mapping: it produces a comprehensive map of the intracellular disease pathway, enabling the identification of a large number of potential protein targets; |
|
·
| |
informed target selection: it provides a variety of different types of targets and information concerning the role each plays in their endogenous state to better select targets more susceptible to pharmaceutical intervention; |
|
·
| |
efficient compound screening: it increases the probability and speed with which compound screening will identify “hits” because it provides detailed knowledge of the target that can be used to guide the design of the compound screen; and |
|
·
| |
risk reduction: it may reduce the risk of failure in the product development process due to serious side effects, including toxicity or other reasons, by selecting only targets that are specific to the disease in question and that have no apparent role in other cell types or signaling pathways. |
Because of the very large numbers of screens employed, our technology is labor intensive. The complexity of our technology requires a high degree of skill and diligence to perform successfully. We believe we have been and will continue to be able to meet these challenges successfully and increase our ability to identify targets for drug discovery.
Pharmacology and Preclinical Development
We believe that the rapid characterization and optimization of compounds identified in high‑throughput screening (HTS) will generate high quality preclinical development candidates. Our pharmacology and preclinical development group facilitates lead optimization by characterizing lead compounds with respect to pharmacokinetics, potency, efficacy and selectivity. The generation of proof‑of‑principle data in animals and the establishment of standard pharmacological models with which to assess lead compounds represent integral components of lead optimization. As programs move through the lead optimization stage, our pharmacology and preclinical development groups support our chemists and biologists by performing the necessary studies, including toxicology, for IND application submissions.
Clinical Development
We have assembled a team of experts in drug development to design and implement clinical trials and to analyze the data derived from these trials. The clinical development group possesses expertise in project management and regulatory affairs. We work with external clinical research organizations with expertise in managing clinical trials, drug formulation, and the manufacture of clinical trial supplies to support our drug development efforts.
Intellectual Property
We are able to protect our technology from unauthorized use by third parties only to the extent that it is covered by valid and enforceable patents or is effectively maintained as a trade secret. Accordingly, patents and other proprietary rights are an essential element of our business. We have about 74 pending patent applications and about 330 issued and active patents in the United States, as well as corresponding pending foreign patent applications and issued foreign patents. Our policy is to file patent applications to protect technology, inventions and improvements to inventions that are commercially important to the development of our business. We seek U.S. and international patent protection for a variety of technologies, including new screening methodologies and other research tools, target molecules that are associated with disease states identified in our screens, and lead compounds that can affect disease pathways. We also intend to seek patent protection or rely upon trade secret rights to protect other technologies that may be used to discover and validate targets and that may be used to identify and develop novel drugs. We seek protection, in part, through confidentiality and proprietary information agreements. We are a party to various license agreements that give us rights to use technologies in our research and development.
Our patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in the various countries where patent protection is obtained. Our material patents relate to compositions of matter covering specific drug candidates in clinical trials that target SYK. These patents will expire, excluding patent term extensions, in 2023, 2024 and 2026. Several of these patents will have patent term extensions, depending on the length of time required to conduct clinical trials.
We currently hold a number of issued patents in the United States, as well as corresponding applications that allow us to pursue patents in other countries, some of which have been allowed and/or granted and others of which we expect to be granted. Specifically, in most cases where we hold a U.S. issued patent, the subject matter is covered at least by an application filed under the Patent Cooperation Treaty (PCT), which is then used or has been used to pursue protection in certain countries that are members of the treaty. Our material patents relate to fostamatinib, an oral SYK inhibitor, and R406, the active metabolite of fostamatinib.
Fostamatinib. Fostamatinib is covered as a composition of matter in a U.S. issued patent that has an expiration date in September 2026, after taking into account a patent term adjustment, and may be granted further protection under the patent term extension rules related to conducting clinical trials. Fostamatinib is also covered under broader composition of matter claims in a U.S. issued patent that has an expiration date in March 2026, after taking into account a patent term adjustment. Methods of using fostamatinib to treat various indications, methods of making fostamatinib, and compositions of matter covering certain intermediates used to make fostamatinib are also covered, respectively, in three U.S. issued patents; the earliest expiration date of any of these patents is in April 2023 and the latest expiration date is in June 2026, after taking into account patent term adjustments. Corresponding applications have been filed in foreign jurisdictions under the PCT, and are at various stages of prosecution. Of note, a patent covering fostamatinib as a composition of matter and in compositions for use treating various diseases has been granted by the European Patent Office.
R406. R406 is covered as a composition of matter in a U.S. issued patent and, with a patent term adjustment, has an expiration date in February 2025. R406 is also covered under two broader composition of matter patents issued in the U.S. expiring in February 2023 and July 2024. Methods of using R406 to treat various indications and compositions of matter covering certain intermediates used to make R406 are also covered under patents described above. Corresponding applications have been filed in foreign jurisdictions under the PCT and are at various stages of prosecution.
Competition
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Many of the drugs that we are attempting to discover will be competing with existing therapies. In addition, a number of companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting. For example, there are existing therapies and drug candidates in development for the treatment of ITP that may be alternative therapies to fostamatinib, if it is ultimately approved for commercialization. We
face, and will continue to face, intense competition from pharmaceutical and biotechnology companies, as well as from academic and research institutions and government agencies, both in the United States and abroad. Some of these competitors are pursuing the development of pharmaceuticals that target the same diseases and conditions as our research programs. Our major competitors include fully integrated pharmaceutical companies that have extensive drug discovery efforts and are developing novel small molecule pharmaceuticals. We also face significant competition from organizations that are pursuing the same or similar technologies, including the discovery of targets that are useful in compound screening, as the technologies used by us in our drug discovery efforts.
Competition may also arise from:
|
·
| |
new or better methods of target identification or validation; |
|
·
| |
other drug development technologies and methods of preventing or reducing the incidence of disease; |
|
·
| |
new small molecules; or |
|
·
| |
other classes of therapeutic agents. |
Our competitors or their collaborative partners may utilize discovery technologies and techniques or partner with collaborators in order to develop products more rapidly or successfully than we or our collaborators are able to do. Many of our competitors, particularly large pharmaceutical companies, have substantially greater financial, technical and human resources and larger research and development staffs than we do. In addition, academic institutions, government agencies and other public and private organizations conducting research may seek patent protection with respect to potentially competitive products or technologies and may establish exclusive collaborative or licensing relationships with our competitors.
We believe that our ability to compete is dependent, in part, upon our ability to create, maintain and license scientifically advanced technology and upon our and our collaborators’ ability to develop and commercialize pharmaceutical products based on this technology, as well as our ability to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary technology or processes and secure sufficient capital resources for the expected substantial time period between technological conception and commercial sales of products based upon our technology. The failure by any of our collaborators or us in any of those areas may prevent the successful commercialization of our potential drug targets.
Many of our competitors, either alone or together with their collaborative partners, have significantly greater experience than we do in:
|
·
| |
identifying and validating targets; |
|
·
| |
screening compounds against targets; and |
|
·
| |
undertaking preclinical testing and clinical trials. |
Accordingly, our competitors may succeed in obtaining patent protection, identifying or validating new targets or discovering new drug compounds before we do.
Our competitors might develop technologies and drugs that are more effective or less costly than any that are being developed by us or that would render our technology and product candidates obsolete and noncompetitive. In addition, our competitors may succeed in obtaining the approval of the FDA or other regulatory agencies for product candidates more rapidly. Companies that complete clinical trials, obtain required regulatory agency approvals and commence commercial sale of their drugs before us may achieve a significant competitive advantage, including certain patent and FDA marketing exclusivity rights that would delay or prevent our ability to market certain products. Any drugs resulting from our research and development efforts, or from our joint efforts with our existing or future
collaborative partners, might not be able to compete successfully with competitors’ existing or future products or obtain regulatory approval in the United States or elsewhere.
We face and will continue to face intense competition from other companies for collaborative arrangements with pharmaceutical and biotechnology companies, for establishing relationships with academic and research institutions and for licenses to additional technologies. These competitors, either alone or with their collaborative partners, may succeed in developing technologies or products that are more effective than ours.
Our ability to compete successfully will depend, in part, on our ability to:
|
·
| |
identify and validate targets; |
|
·
| |
discover candidate drug compounds that interact with the targets we identify; |
|
·
| |
attract and retain scientific and product development personnel; |
|
·
| |
obtain patent or other proprietary protection for our new drug compounds and technologies; and |
|
·
| |
enter commercialization agreements for our new drug compounds. |
Research and Development Expenses
A significant portion of our operating expenses is related to research and development and we intend to maintain our strong commitment to research and development. See “Item 8. Financial Statements and Supplementary Data” of this Annual Report on Form 10‑K for costs and expenses related to research and development, and other financial information for each of the fiscal years 2015, 2014 and 2013.
Government Regulation
Our ongoing development activities are and will continue to be subject to extensive regulation by numerous governmental authorities in the United States and other countries, including the FDA under the Federal Food, Drug and Cosmetic Act. The regulatory review and approval process is expensive and uncertain. Securing FDA approval requires the submission of extensive preclinical and clinical data and supporting information to the FDA for each indication to establish a product candidate’s safety and efficacy.
Preclinical studies generally are conducted in laboratory animals to evaluate the potential safety and the efficacy of a product. Drug developers submit the results of preclinical studies to the FDA as part of an IND application that must be approved before clinical trials can begin in humans. Typically, clinical evaluation involves a time consuming and costly three‑phase process.
|
·
| |
Phase 1—Clinical trials are conducted with a small number of patients to determine the early safety profile, maximum tolerated dose and pharmacological properties of the product in human volunteers. |
|
·
| |
Phase 2—Clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. |
|
·
| |
Phase 3—Large‑scale, multi‑center, comparative clinical trials are conducted with patients afflicted with a specific disease in order to determine safety and efficacy as primary support for regulatory approval by the FDA to market a product candidate for a specific disease. |
The approval process takes many years, requires the expenditure of substantial resources and may involve ongoing requirements for post‑ marketing studies. Clinical trials are subject to oversight by institutional review boards and the FDA. In addition, clinical trials:
|
·
| |
must be conducted in conformance with the FDA’s good clinical practices and other applicable regulations; |
|
·
| |
must meet requirements for institutional review board oversight; |
|
·
| |
must meet requirements for informed consent; |
|
·
| |
are subject to continuing FDA oversight; |
|
·
| |
may require large numbers of participants; and |
|
·
| |
may be suspended by us, our collaborators or the FDA at any time if it is believed that the subjects participating in these trials are being exposed to unacceptable health risks or if the FDA finds deficiencies in the IND or the conduct of these trials. |
Even if we are able to achieve success in our clinical testing, we, or our collaborative partners, must provide the FDA and foreign regulatory authorities with clinical data that demonstrates the safety and efficacy of our products in humans before they can be approved for commercial sale. We do not know whether any current or future clinical trials will demonstrate sufficient safety and efficacy necessary to obtain the requisite regulatory approvals or will result in marketable products. Our failure, or the failure of our strategic partners, to adequately demonstrate the safety and efficacy of our products under development will prevent receipt of FDA and similar foreign regulatory approval and, ultimately, commercialization of our products.
Any clinical trial may fail to produce results satisfactory to the FDA. Preclinical and clinical data can be interpreted in different ways, which could delay, limit or prevent regulatory approval. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be repeated or a program to be terminated. In addition, delays or rejections may be encountered based upon additional government regulation from future legislation or administrative action or changes in FDA policy or interpretation during the period of product development, clinical trials and FDA regulatory review. Failure to comply with applicable FDA or other applicable regulatory requirements may result in criminal prosecution, civil penalties, recall or seizure of products, total or partial suspension of production or injunction, as well as other regulatory action against our potential products, collaborative partners or us.
Outside the United States, our ability to market a product is contingent upon receiving a marketing authorization from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. At present, foreign marketing authorizations are applied for at a national level, although within the E.U., registration procedures are available to companies wishing to market a product in more than one E.U. member state. If the regulatory authority is satisfied that adequate evidence of safety, quality and efficacy has been presented, a marketing authorization will be granted. This foreign regulatory approval process involves all of the risks associated with FDA clearance.
Manufacturing and Raw Materials
We currently rely on, and will continue to rely on, third party contract manufacturers to produce sufficient quantities of our product candidates for use in our preclinical and clinical trials.
Employees
As of December 31, 2015, we had 126 employees. None of our employees are represented by a collective bargaining arrangement, and we believe our relationship with our employees is good. Recruiting and retaining qualified scientific personnel to perform research and development work in the future will be critical to our success. We may not be able to attract and retain personnel on acceptable terms given the competition among pharmaceutical and biotechnology companies, academic and research institutions and government agencies for experienced scientists.
Scientific and Medical Advisors
We utilize scientists and physicians to advise us on scientific and medical matters as part of our ongoing research and product development efforts, including experts in clinical trial design, preclinical development work, chemistry, biology, immunology, muscle wasting and metabolism, general metabolism and oncology. Certain of our consultants receive non‑employee options to purchase our common stock and certain of our scientific and medical advisors receive honorarium for time spent assisting us.
Available Information
Our website is located at www.rigel.com. The information found on our website is not part of or incorporated by reference into this Annual Report on Form 10‑K. We electronically file with the Securities and Exchange Commission (SEC) our Annual Report on Form 10‑K, Quarterly Reports on Form 10‑Q, Current Reports on Form 8‑K and amendments to the reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act. We make available free of charge on or through our website copies of these reports as soon as reasonably practicable after we electronically file these reports with, or furnish them to, the SEC. Further, copies of these reports are available at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the SEC at 1‑800‑SEC‑0330. The SEC also maintains an internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC at www.sec.gov.
Item 1A. Risk Factors
In evaluating our business, you should carefully consider the following risks, as well as the other information contained in this Annual Report on Form 10‑K. These risk factors could cause our actual results to differ materially from those contained in forward‑looking statements we have made in this Annual Report on Form 10‑K and those we may make from time to time. If any of the following risks actually occurs, our business, financial condition and operating results could be harmed. The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us, or that we currently see as immaterial, may also harm our business.
We will need additional capital in the future to sufficiently fund our operations and research.
We have consumed substantial amounts of capital to date as we continue our research and development activities, including preclinical studies and clinical trials. We initiated a Phase 3 clinical program to study fostamatinib in ITP in July 2014 on our own, which may accelerate our need for additional capital. We may seek another collaborator or licensee in the future for further clinical development and commercialization of fostamatinib, as well as our other clinical programs, which we may not be able to obtain on commercially reasonable terms or at all. We believe that our existing capital resources will be sufficient to support our current and projected funding requirements into the third quarter of 2017. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development of our product candidates and other research and development activities, including risks and uncertainties that could impact the rate of progress of our development activities, we are unable to estimate with certainty the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical trials and other research and development activities. We will continue to need additional capital and the amount of future capital needed will depend largely on the success of our internally developed programs as they proceed in later and more expensive clinical trials, including any additional clinical trials that we may decide to conduct with respect to fostamatinib. Unless and until we are able to generate a sufficient amount of product, royalty or milestone revenue, which may never occur, we expect to finance future cash needs through public and/or private offerings of equity securities, debt financings or collaboration and licensing arrangements, as well as through interest income earned on the investment of our cash balances and short-term investments. With the exception of contingent and royalty payments that we may receive under our existing collaborations, we do not currently have any commitments for future funding. We do not know whether additional financing will be available when needed, or that, if available, we will obtain financing on reasonable terms.
To the extent we raise additional capital by issuing equity securities in the future, our stockholders could at that time experience substantial dilution. Any debt financing that we are able to obtain may involve operating covenants that restrict our business. To the extent that we raise additional funds through any new collaboration and licensing arrangements, we may be required to relinquish some rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us.
Our future funding requirements will depend on many uncertain factors.
Our future funding requirements will depend upon many factors, many of which are beyond our control, including, but not limited to:
|
·
| |
the progress and success of our clinical trials and preclinical activities (including studies and manufacture of materials) of our product candidates conducted by us; |
|
·
| |
the progress of research and development programs carried out by us; |
|
·
| |
any changes in the breadth of our research and development programs; |
|
·
| |
the ability to achieve the events identified in our collaborative agreements that may trigger payments to us from our collaboration partners; |
|
·
| |
the progress of the research and development efforts of our collaborative partners; |
|
·
| |
our ability to acquire or license other technologies or compounds that we seek to pursue; |
|
·
| |
our ability to manage our growth; |
|
·
| |
competing technological and market developments; |
|
·
| |
the costs and timing of obtaining, enforcing and defending our patent and other intellectual property rights; |
|
·
| |
the costs and timing of regulatory filings and approvals by us and our collaborators; and |
|
·
| |
expenses associated with any unforeseen litigation, including any securities class action lawsuits. |
Insufficient funds may require us to delay, scale back or eliminate some or all of our research and development programs, to lose rights under existing licenses or to relinquish greater or all rights to product candidates at an earlier stage of development or on less favorable terms than we would otherwise choose or may adversely affect our ability to operate as a going concern.
There is a high risk that drug discovery and development efforts might not generate successful product candidates.
At the present time, the majority of our operations are in various stages of drug identification and development. We currently have four product candidates in the clinical testing stage. In our industry, it is statistically unlikely that the limited number of compounds that we have identified as potential product candidates will actually lead to successful product development efforts, and we do not expect any drugs resulting from our research to be commercially available for several years, if at all.
Our compounds in clinical trials and our future leads for potential drug compounds are subject to the risks and failures inherent in the development of pharmaceutical products. These risks include, but are not limited to, the inherent difficulty in selecting the right drug and drug target and avoiding unwanted side effects, as well as unanticipated problems relating to product development, testing, enrollment, obtaining regulatory approvals, maintaining regulatory compliance, manufacturing, competition and costs and expenses that may exceed current estimates. In future clinical trials, we or our partners may discover additional side effects and/or higher frequency of side effects than those observed in completed clinical trials. The results of preliminary and mid-stage clinical trials do not necessarily predict clinical or commercial success, and larger later-stage clinical trials may fail to confirm the results observed in the previous clinical trials. Similarly, a clinical trial may show that a product candidate is safe and effective for certain patient populations in a particular indication, but other clinical trials may fail to confirm those results in a subset of that population or in a different patient population, which may limit the potential market for that product candidate. With respect to our own compounds in development, we have established anticipated timelines with respect to the initiation of clinical trials
based on existing knowledge of the compounds. However, we cannot provide assurance that we will meet any of these timelines for clinical development. Additionally, the initial results of the completed Phase 2 clinical trial of fostamatinib in ITP do not necessarily predict final results and the results may not be repeated in later clinical trials.
Because of the uncertainty of whether the accumulated preclinical evidence (pharmacokinetic, pharmacodynamic, safety and/or other factors) or early clinical results will be observed in later clinical trials, we can make no assurances regarding the likely results from our future clinical trials or the impact of those results on our business.
We might not be able to commercialize our product candidates successfully if problems arise in the clinical testing and approval process.
Commercialization of our product candidates depends upon successful completion of extensive preclinical studies and clinical trials to demonstrate their safety and efficacy for humans. Preclinical testing and clinical development are long, expensive and uncertain processes.
In connection with clinical trials of our product candidates, we face the risks that:
|
·
| |
the product candidate may not prove to be effective; |
|
·
| |
the product candidate may cause harmful side effects; |
|
·
| |
the clinical results may not replicate the results of earlier, smaller trials; |
|
·
| |
we, or the FDA or similar foreign regulatory authorities, may terminate or suspend the trials; |
|
·
| |
our results may not be statistically significant; |
|
·
| |
patient recruitment and enrollment may be slower than expected; |
|
·
| |
patients may drop out of the trials; and |
|
·
| |
regulatory and clinical trial requirements, interpretations or guidance may change. |
We do not know whether we will be permitted to undertake clinical trials of potential products beyond the trials already concluded and the trials currently in process. It will take us, or our collaborative partners several years to complete any such testing, and failure can occur at any stage of testing. Interim results of trials do not necessarily predict final results, and acceptable results in early trials may not be repeated in later trials. A number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in advanced clinical trials, even after achieving promising results in earlier trials. For example, R348, our topical ophthalmic JAK/SYK inhibitor, did not meet the primary or secondary endpoints in a completed Phase 2 clinical trial in patients with dry eye disease. Moreover, we or our collaborative partners or regulators may decide to discontinue development of any or all of these projects at any time for commercial, scientific or other reasons. For example, in August 2014, we have discontinued our indirect AMPK activator program, R118, due to its side-effect profile in Phase 1 clinical trials.
We initiated a Phase 3 clinical program to study fostamatinib in ITP in July 2014 on our own. We cannot assure you that we will be able to successfully complete the clinical development of fostamatinib or receive regulatory approval to ultimately commercialize fostamatinib. If we are unable to complete the clinical development of fostamatinib, our business will be harmed.
Delays in clinical testing could result in increased costs to us.
We may not be able to initiate or continue clinical studies or trials for our product candidates if we are unable to locate and enroll a sufficient number of eligible patients to participate in these clinical trials as required by the FDA or
other regulatory authorities. Even if we are able to enroll a sufficient number of patients in our clinical trials, if the pace of enrollment is slower than we expect, the development costs for our product candidates may increase and the completion of our clinical trials may be delayed or our clinical trials could become too expensive to complete. Significant delays in clinical testing could materially impact our product development costs and timing. For example, in July 2014, we initiated our Phase 3 clinical program to study fostamatinib in ITP, in which a total of 150 ITP patients will be randomized into two identical multi-center, double-blind, placebo-controlled clinical trials. Our estimates regarding timing are based on a number of assumptions, including assumptions based on past experience with our other clinical programs. We completed patient enrollment of the first trial in January 2016. If we are unable to enroll the patients in the second trial at the projected rate, the completion of the clinical program could be delayed and the costs of conducting the program could increase, either of which could harm our business.
Clinical trials can be delayed for a variety of reasons, including delays in obtaining regulatory approval to commence a study, delays from scaling up of a study, delays in reaching agreement on acceptable clinical trial agreement terms with prospective clinical sites, delays in obtaining institutional review board approval to conduct a study at a prospective clinical site or delays in recruiting subjects to participate in a study. In addition, we typically rely on third-party clinical investigators to conduct our clinical trials and other third-party organizations to oversee the operations of such trials and to perform data collection and analysis. The clinical investigators are not our employees, and we cannot control the amount or timing of resources that they devote to our programs. Failure of the third-party organizations to meet their obligations could adversely affect clinical development of our products. As a result, we may face additional delaying factors outside our control if these parties do not perform their obligations in a timely fashion. While we have not yet experienced delays that have materially impacted our clinical trials or product development costs, delays of this sort could occur for the reasons identified above or other reasons. If we have delays in testing or obtaining regulatory approvals, our product development costs will increase. For example, we may need to make additional payments to third-party investigators and organizations to retain their services or we may need to pay recruitment incentives. If the delays are significant, our financial results and the commercial prospects for our product candidates will be harmed, and our ability to become profitable will be delayed. Moreover, these third-party investigators and organizations may also have relationships with other commercial entities, some of which may compete with us. If these third-party investigators and organizations assist our competitors at our expense, it could harm our competitive position.
We have obtained orphan drug designation from the FDA for fostamatinib for the treatment of ITP, but we may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug or biologic intended to treat a rare disease or condition, which is defined as one occurring in a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug or biologic will be recovered from sales in the United States. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full Biologics License Application, or BLA, to market the same biologic for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or where the manufacturer is unable to assure sufficient product quantity.
Even though we have received orphan drug designation for fostamatinib for the treatment of ITP, we may not be the first to obtain marketing approval for the orphan-designated indication due to the uncertainties associated with developing pharmaceutical products. In addition, exclusive marketing rights in the United States may be limited if we seek approval for an indication broader than the orphan-designated indication or may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Further, even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs with different active moieties can be approved for the same condition. Even after an orphan product is approved, the FDA can subsequently approve the same drug with the same active moiety for the same condition if the FDA
concludes that the later drug is safer, more effective, or makes a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
Our success as a company is uncertain due to our history of operating losses and the uncertainty of any future profitability.
We incurred a loss from operations of approximately $51.7 million for the year ended December 31, 2015. Other than for 2010, we have historically incurred losses from operations each year since we were incorporated in June 1996, due in large part to the significant research and development expenditures required to identify and validate new product candidates and pursue our development efforts. We expect to continue to incur losses from operations and there can be no assurance that we will generate operating income in the foreseeable future. Currently, our only potential sources of revenues are upfront payments, research and development contingent payments and royalty payments pursuant to our collaboration arrangements, which may never materialize if our collaborators do not achieve certain events or generate net sales to which these contingent payments are dependent on. If our drug candidates fail or do not gain regulatory approval, or if our drugs do not achieve market acceptance, we may not be profitable. As of December 31, 2015, we had an accumulated deficit of approximately $991.6 million. The extent of our future losses or profitability, if any, is highly uncertain.
If our corporate collaborations or license agreements are unsuccessful, or if we fail to form new corporate collaborations or license agreements, our research and development efforts could be delayed.
Our strategy depends upon the formation and sustainability of multiple collaborative arrangements and license agreements with third parties now and in the future. We rely on these arrangements for not only financial resources, but also for expertise we need now and in the future relating to clinical trials, manufacturing, sales and marketing, and for licenses to technology rights. To date, we have entered into several such arrangements with corporate collaborators; however, we do not know if these collaborations or additional collaborations with third parties, if any, will dedicate sufficient resources or if any development or commercialization efforts by third parties will be successful. In addition, our corporate collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a drug candidate or development program. Should a collaborative partner fail to develop or commercialize a compound or product to which it has rights from us for any reason, including corporate restructuring, such failure might delay our ongoing research and development efforts, because we might not receive any future payments, and we would not receive any royalties associated with such compound or product. We initiated a Phase 3 clinical program to study fostamatinib in ITP in July 2014 on our own. We may seek another collaborator or licensee in the future for clinical development and commercialization of fostamatinib, as well as our other clinical programs, which we may not be able to obtain on commercially reasonable terms or at all. If we are unable to form new collaborations or enter into new license agreements, our research and development efforts could be delayed. In addition, the continuation of some of our partnered drug discovery and development programs may be dependent on the periodic renewal of our corporate collaborations.
Each of our collaborations could be terminated by the other party at any time, and we may not be able to renew these collaborations on acceptable terms, if at all, or negotiate additional corporate collaborations on acceptable terms, if at all. If these collaborations terminate or are not renewed, any resultant loss of revenues from these collaborations or loss of the resources and expertise of our collaborative partners could adversely affect our business.
Conflicts also might arise with collaborative partners concerning proprietary rights to particular compounds. While our existing collaborative agreements typically provide that we retain milestone payments and royalty rights with respect to drugs developed from certain derivative compounds, any such payments or royalty rights may be at reduced rates, and disputes may arise over the application of derivative payment provisions to such drugs, and we may not be successful in such disputes. Additionally, the management teams of our collaborators may change for various reasons including due to being acquired. Different management teams or an acquiring company of our collaborators may have different priorities which may have adverse results on the collaboration with us.
We are also a party to various license agreements that give us rights to use specified technologies in our
research and development processes. The agreements pursuant to which we have in-licensed technology permit our licensors to terminate the agreements under certain circumstances. If we are not able to continue to license these and future technologies on commercially reasonable terms, our product development and research may be delayed or otherwise adversely affected.
If conflicts arise between our collaborators or advisors and us, any of them may act in their self-interest, which may be adverse to our stockholders’ interests.
If conflicts arise between us and our corporate collaborators or scientific advisors, the other party may act in its self-interest and not in the interest of our stockholders. Some of our corporate collaborators are conducting multiple product development efforts within each disease area that is the subject of the collaboration with us or may be acquired or merged with a company having a competing program. In some of our collaborations, we have agreed not to conduct, independently or with any third party, any research that is competitive with the research conducted under our collaborations. Our collaborators, however, may develop, either alone or with others, products in related fields that are competitive with the products or potential products that are the subject of these collaborations. Competing products, either developed by our collaborators or to which our collaborators have rights, may result in their withdrawal of support for our product candidates.
If any of our corporate collaborators were to breach or terminate its agreement with us or otherwise fail to conduct the collaborative activities successfully and in a timely manner, the preclinical or clinical development or commercialization of the affected product candidates or research programs could be delayed or terminated. We generally do not control the amount and timing of resources that our corporate collaborators devote to our programs or potential products. We do not know whether current or future collaborative partners, if any, might pursue alternative technologies or develop alternative products either on their own or in collaboration with others, including our competitors, as a means for developing treatments for the diseases targeted by collaborative arrangements with us.
If we are unable to obtain regulatory approval to market products in the United States and foreign jurisdictions, we will not be permitted to commercialize products we or our collaborative partners may develop.
We cannot predict whether regulatory clearance will be obtained for any product that we, or our collaborative partners, hope to develop. Satisfaction of regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the product and requires the expenditure of substantial resources. Of particular significance to us are the requirements relating to research and development and testing.
Before commencing clinical trials in humans in the United States, we, or our collaborative partners, will need to submit and receive approval from the FDA of an IND. Clinical trials are subject to oversight by institutional review boards and the FDA and:
|
·
| |
must be conducted in conformance with the FDA’s good clinical practices and other applicable regulations; |
|
·
| |
must meet requirements for institutional review board oversight; |
|
·
| |
must meet requirements for informed consent; |
|
·
| |
are subject to continuing FDA and regulatory oversight; |
|
·
| |
may require large numbers of test subjects; and |
|
·
| |
may be suspended by us, our collaborators or the FDA at any time if it is believed that the subjects participating in these trials are being exposed to unacceptable health risks or if the FDA finds deficiencies in the IND or the conduct of these trials. |
While we have stated that we intend to file additional INDs for future product candidates, this is only a statement of intent, and we may not be able to do so because we may not be able to identify potential product candidates.
In addition, the FDA may not approve any IND in a timely manner, or at all.
Before receiving FDA approval to market a product, we must demonstrate with substantial clinical evidence that the product is safe and effective in the patient population and the indication that will be treated. Data obtained from preclinical and clinical activities are susceptible to varying interpretations that could delay, limit or prevent regulatory approvals. In addition, delays or rejections may be encountered based upon additional government regulation from future legislation or administrative action or changes in FDA policy during the period of product development, clinical trials and FDA regulatory review. Failure to comply with applicable FDA or other applicable regulatory requirements may result in criminal prosecution, civil penalties, recall or seizure of products, total or partial suspension of production or injunction, adverse publicity, as well as other regulatory action against our potential products or us. Additionally, we have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approval.
If regulatory approval of a product is granted, this approval will be limited to those indications or disease states and conditions for which the product is demonstrated through clinical trials to be safe and efficacious. We cannot assure you that any compound developed by us, alone or with others, will prove to be safe and efficacious in clinical trials and will meet all of the applicable regulatory requirements needed to receive marketing approval.
Outside the United States, our ability, or that of our collaborative partners, to market a product is contingent upon receiving a marketing authorization from the appropriate regulatory authorities. This foreign regulatory approval process typically includes all of the risks and costs associated with FDA approval described above and may also include additional risks and costs, such as the risk that such foreign regulatory authorities, which often have different regulatory and clinical trial requirements, interpretations and guidance from the FDA, may require additional clinical trials or results for approval of a product candidate, any of which could result in delays, significant additional costs or failure to obtain such regulatory approval. For example, there can be no assurance that we or our collaborative partners will not have to provide additional information or analysis, or conduct additional clinical trials, before receiving approval to market product candidates.
Our success is dependent on intellectual property rights held by us and third parties, and our interest in such rights is complex and uncertain.
Our success will depend to a large part on our own, our licensees’ and our licensors’ ability to obtain and defend patents for each party’s respective technologies and the compounds and other products, if any, resulting from the application of such technologies. We have about 74 pending patent applications and about 330 issued and active patents in the United States, as well as corresponding pending foreign patent applications and issued foreign patents. In the future, our patent position might be highly uncertain and involve complex legal and factual questions. For example, we may be involved in post-grant proceedings before the United States Patent and Trademark Office. Post-grant proceedings are complex and expensive legal proceedings and there is no assurance we will be successful in any such proceedings. A post-grant proceeding could result in our losing our patent rights and/or our freedom to operate and/or require us to pay significant royalties. Additional uncertainty may result because no consistent policy regarding the breadth of legal claims allowed in biotechnology patents has emerged to date. Accordingly, we cannot predict the breadth of claims allowed in our or other companies’ patents.
Because the degree of future protection for our proprietary rights is uncertain, we cannot assure you that:
|
·
| |
we were the first to make the inventions covered by each of our pending patent applications; |
|
·
| |
we were the first to file patent applications for these inventions; |
|
·
| |
others will not independently develop similar or alternative technologies or duplicate any of our technologies; |
|
·
| |
any of our pending patent applications will result in issued patents; |
|
·
| |
any patents issued to us or our collaborators will provide a basis for commercially-viable products or will provide us with any competitive advantages or will not be challenged by third parties; |
|
·
| |
we will develop additional proprietary technologies that are patentable; or |
|
·
| |
the patents of others will not have a negative effect on our ability to do business. |
We rely on trade secrets to protect technology where we believe patent protection is not appropriate or obtainable; however, trade secrets are difficult to protect. While we require employees, collaborators and consultants to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information.
We are a party to certain in-license agreements that are important to our business, and we generally do not control the prosecution of in-licensed technology. Accordingly, we are unable to exercise the same degree of control over this intellectual property as we exercise over our internally-developed technology. Moreover, some of our academic institution licensors, research collaborators and scientific advisors have rights to publish data and information in which we have rights. If we cannot maintain the confidentiality of our technology and other confidential information in connection with our collaborations, our ability to receive patent protection or protect our proprietary information may otherwise be impaired. In addition, some of the technology we have licensed relies on patented inventions developed using U.S. government resources.
The U.S. government retains certain rights, as defined by law, in such patents, and may choose to exercise such rights. Certain of our in-licenses may be terminated if we fail to meet specified obligations. If we fail to meet such obligations and any of our licensors exercise their termination rights, we could lose our rights under those agreements. If we lose any of our rights, it may adversely affect the way we conduct our business. In addition, because certain of our licenses are sublicenses, the actions of our licensors may affect our rights under those licenses.
If a dispute arises regarding the infringement or misappropriation of the proprietary rights of others, such dispute could be costly and result in delays in our research and development activities and partnering.
Our success will depend, in part, on our ability to operate without infringing or misappropriating the proprietary rights of others. There are many issued patents and patent applications filed by third parties relating to products or processes that are similar or identical to our licensors or ours, and others may be filed in the future. There may also be copyrights or trademarks that third parties hold. There can be no assurance that our activities, or those of our licensors, will not violate intellectual property rights of others. We believe that there may be significant litigation in the industry regarding patent and other intellectual property rights, and we do not know if our collaborators or we would be successful in any such litigation. Any legal action against our collaborators or us claiming damages or seeking to enjoin commercial activities relating to the affected products, our methods or processes could:
|
·
| |
require our collaborators or us to obtain a license to continue to use, manufacture or market the affected products, methods or processes, which may not be available on commercially reasonable terms, if at all; |
|
·
| |
prevent us from using the subject matter claimed in the patents held by others; |
|
·
| |
subject us to potential liability for damages; |
|
·
| |
consume a substantial portion of our managerial and financial resources; and |
|
·
| |
result in litigation or administrative proceedings that may be costly, whether we win or lose. |
Our research and development efforts will be seriously jeopardized if we are unable to attract and retain key employees and relationships.
As a small company, our success depends on the continued contributions of our principal management and
scientific personnel and on our ability to develop and maintain important relationships with leading academic institutions, scientists and companies in the face of intense competition for such personnel. In particular, our research programs depend on our ability to attract and retain highly skilled chemists, other scientists, and development, regulatory and clinical personnel. If we lose the services of any of our key personnel, our research and development efforts could be seriously and adversely affected. Our employees can terminate their employment with us at any time.
Our ability to use net operating losses and certain other tax attributes is uncertain and may be limited.
Our ability to use our federal and state net operating losses to offset potential future taxable income and related income taxes that would otherwise be due is dependent upon our generation of future taxable income before the expiration dates of the net operating losses, and we cannot predict with certainty when, or whether, we will generate sufficient taxable income to use all of our net operating losses. In addition, utilization of net operating losses to offset potential future taxable income and related income taxes that would otherwise be due is subject to annual limitations under the “ownership change” provisions of Sections 382 and 383 of the Internal Revenue Code of 1986, as amended (Internal Revenue Code) and similar state provisions, which may result in the expiration of net operating losses before future utilization. In general, under the Code, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating losses and other pre-change tax attributes (such as research and development credit carryforwards) to offset its post-change taxable income or taxes may be limited. Our equity offerings and other changes in our stock ownership, some of which are outside of our control, may have resulted or could in the future result in an ownership change. Although we have completed studies to provide reasonable assurance that an ownership change limitation would not apply, we cannot be certain that a taxing authority would reach the same conclusion. If, after a review or audit, an ownership change limitation were to apply, utilization of our domestic net operating losses and tax credit carryforwards could be limited in future periods and a portion of the carryforwards could expire before being available to reduce future income tax liabilities.
Because we expect to be dependent upon collaborative and license agreements, we might not meet our strategic objectives.
Our ability to generate revenue in the near term depends on the timing of recognition of certain upfront payments, achievement of certain payment triggering events with our existing collaboration agreements and our ability to enter into additional collaborative agreements with third parties. Our ability to enter into new collaborations and the revenue, if any, that may be recognized under these collaborations is highly uncertain. If we are unable to enter into one or more new collaborations, our business prospects could be harmed, which could have an immediate adverse effect on our ability to continue to develop our compounds and on the trading price of our stock. Our ability to enter into a collaboration may be dependent on many factors, such as the results of our clinical trials, competitive factors and the fit of one of our programs with another company’s risk tolerance, including toward regulatory issues, patent portfolio, clinical pipeline, the stage of the available data, particularly if it is early, overall corporate goals and financial position.
To date, a portion of our revenues have been related to the research or transition phase of each of our collaborative agreements. Such revenues are for specified periods, and the impact of such revenues on our results of operations is at least partially offset by corresponding research costs. Following the completion of the research or transition phase of each collaborative agreement, additional revenues may come only from payments triggered by milestones and/or the achievement of other contingent events, and royalties, which may not be paid, if at all, until certain conditions are met. This risk is heightened due to the fact that unsuccessful research efforts may preclude us from receiving any contingent payments under these agreements. Our receipt of revenues from collaborative arrangements is also significantly affected by the timing of efforts expended by us and our collaborators and the timing of lead compound identification. We have received payments from our collaborations with Aclaris, BMS, AZ, BerGenBio, Janssen Pharmaceutica N.V., a division of Johnson & Johnson, Novartis Pharma A.G., Daiichi, Merck & Co., Inc., Merck Serono and Pfizer. Under many agreements, future payments may not be earned until the collaborator has advanced product candidates into clinical testing, which may never occur or may not occur until some time well into the future. If we are not able to generate revenue under our collaborations when and in accordance with our expectations or the expectations of industry analysts, this failure could harm our business and have an immediate adverse effect on the trading price of our common stock.
Our business requires us to generate meaningful revenue from royalties and licensing agreements. To date, we have not received any revenue from royalties for the commercial sale of drugs, and we do not know when we will receive any such revenue, if at all.
Securities class action lawsuits or other litigation could result in substantial damages and may divert management’s time and attention from our business.
We have been subject to class action lawsuits in the past, including a securities class action lawsuit commenced in the United States District Court for the Northern District of California in February 2009, that was ultimately dismissed in November 2012. However, we may be subject to similar or completely unrelated claims in the future, such as those that might occur if there was to be a change in our corporate strategy. These and other lawsuits are subject to inherent uncertainties, and the actual costs to be incurred relating to the lawsuit will depend upon many unknown factors. The outcome of litigation is necessarily uncertain, and we could be forced to expend significant resources in the defense of such suits, and we may not prevail. Monitoring and defending against legal actions is time-consuming for our management and detracts from our ability to fully focus our internal resources on our business activities. In addition, we may incur substantial legal fees and costs in connection with any such litigation. We have not established any reserves for any potential liability relating to any such potential lawsuits. It is possible that we could, in the future, incur judgments or enter into settlements of claims for monetary damages. A decision adverse to our interests on any such actions could result in the payment of substantial damages, or possibly fines, and could have a material adverse effect on our cash flow, results of operations and financial position.
We lack the capability to manufacture compounds for development and rely on third parties to manufacture our product candidates, and we may be unable to obtain required material in a timely manner, at an acceptable cost or at a quality level required to receive regulatory approval.
We currently do not have the manufacturing capabilities or experience necessary to produce our product candidates for clinical trials, including fostamatinib for ITP, IgAN and AIHA, and R348 for dry eye in GvHD. For each clinical trial of our unpartnered product candidates, we rely on third-party manufacturers for the active pharmaceutical ingredients, as well as various manufacturers to manufacture starting components, excipients and formulated drug products. We rely on manufacturers to produce and deliver all of the materials required for our clinical trials, and many of our preclinical efforts, on a timely basis and to comply with applicable regulatory requirements, including the FDA’s current Good Manufacturing Practices (cGMP). In addition, we rely on our suppliers to deliver sufficient quantities of materials produced under cGMP conditions to enable us to conduct planned preclinical studies and clinical trials.
Our current and anticipated future dependence upon these third-party manufacturers may adversely affect our ability to develop and commercialize product candidates on a timely and competitive basis. These manufacturers may not be able to produce material on a timely basis or manufacture material at the quality level or in the quantity required to meet our development timelines and applicable regulatory requirements and may also experience a shortage in qualified personnel. We may not be able to maintain or renew our existing third-party manufacturing arrangements, or enter into new arrangements, on acceptable terms, or at all. Our third party manufacturers could terminate or decline to renew our manufacturing arrangements based on their own business priorities, at a time that is costly or inconvenient for us. If we are unable to contract for the production of materials in sufficient quantity and of sufficient quality on acceptable terms, our planned clinical trials may be significantly delayed. Manufacturing delays could postpone the filing of our IND applications and/or the initiation or completion of clinical trials that we have currently planned or may plan in the future.
Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the Drug Enforcement Administration, and other federal and state agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards and they may not be able to comply. Switching manufacturers may be difficult because the number of potential manufacturers is limited. It may be difficult or impossible for us to find a replacement manufacturer quickly on acceptable terms, or at all. Additionally, if we are required to enter into new supply arrangements, we may not be able to obtain approval from the FDA of any alternate supplier in a timely manner, or at
all, which could delay or prevent the clinical development and commercialization of any related product candidates. Failure of our third-party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on us, including fines, civil penalties, delays in or failure to grant marketing approval of our product candidates, injunctions, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products and compounds, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our business.
If our competitors develop technologies that are more effective than ours, our commercial opportunity will be reduced or eliminated.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Many of the drugs that we are attempting to discover will be competing with existing therapies. In addition, a number of companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting. For example, there are existing therapies and drug candidates in development for the treatment of ITP that may be alternative therapies to fostamatinib, if it is ultimately approved for commercialization. We face, and will continue to face, intense competition from pharmaceutical and biotechnology companies, as well as from academic and research institutions and government agencies, both in the United States and abroad. Some of these competitors are pursuing the development of pharmaceuticals that target the same diseases and conditions as our research programs. Our major competitors include fully integrated pharmaceutical companies that have extensive drug discovery efforts and are developing novel small-molecule pharmaceuticals. We also face significant competition from organizations that are pursuing the same or similar technologies, including the discovery of targets that are useful in compound screening, as the technologies used by us in our drug discovery efforts.
Competition may also arise from:
|
·
| |
new or better methods of target identification or validation; |
|
·
| |
other drug development technologies and methods of preventing or reducing the incidence of disease; |
|
·
| |
new small molecules; or |
|
·
| |
other classes of therapeutic agents. |
Our competitors or their collaborative partners may utilize discovery technologies and techniques or partner with collaborators in order to develop products more rapidly or successfully than we or our collaborators are able to do. Many of our competitors, particularly large pharmaceutical companies, have substantially greater financial, technical and human resources and larger research and development staffs than we do. In addition, academic institutions, government agencies and other public and private organizations conducting research may seek patent protection with respect to potentially competitive products or technologies and may establish exclusive collaborative or licensing relationships with our competitors.
We believe that our ability to compete is dependent, in part, upon our ability to create, maintain and license scientifically-advanced technology and upon our and our collaborators’ ability to develop and commercialize pharmaceutical products based on this technology, as well as our ability to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary technology or processes and secure sufficient capital resources for the expected substantial time period between technological conception and commercial sales of products based upon our technology. The failure by any of our collaborators or us in any of those areas may prevent the successful commercialization of our potential drug targets.
Many of our competitors, either alone or together with their collaborative partners, have significantly greater experience than we do in:
|
·
| |
identifying and validating targets; |
|
·
| |
screening compounds against targets; and |
|
·
| |
undertaking preclinical testing and clinical trials. |
Accordingly, our competitors may succeed in obtaining patent protection, identifying or validating new targets or discovering new drug compounds before we do.
Our competitors might develop technologies and drugs that are more effective or less costly than any that are being developed by us or that would render our technology and product candidates obsolete and noncompetitive. In addition, our competitors may succeed in obtaining the approval of the FDA or other regulatory agencies for product candidates more rapidly. Companies that complete clinical trials, obtain required regulatory agency approvals and commence commercial sale of their drugs before us may achieve a significant competitive advantage, including certain patent and FDA marketing exclusivity rights that would delay or prevent our ability to market certain products. Any drugs resulting from our research and development efforts, or from our joint efforts with our existing or future collaborative partners, might not be able to compete successfully with competitors’ existing or future products or obtain regulatory approval in the United States or elsewhere.
We face and will continue to face intense competition from other companies for collaborative arrangements with pharmaceutical and biotechnology companies, for establishing relationships with academic and research institutions and for licenses to additional technologies. These competitors, either alone or with their collaborative partners, may succeed in developing technologies or products that are more effective than ours.
Our stock price may be volatile, and our stockholders’ investment in our stock could decline in value.
The market prices for our common stock and the securities of other biotechnology companies have been highly volatile and may continue to be highly volatile in the future. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
|
·
| |
the progress and success of our clinical trials and preclinical activities (including studies and manufacture of materials) of our product candidates conducted by us; |
|
·
| |
the receipt or failure to receive the additional funding necessary to conduct our business; |
|
·
| |
selling by large stockholders; |
|
·
| |
presentations of detailed clinical trial data at medical and scientific conferences and investor perception thereof; |
|
·
| |
announcements of technological innovations or new commercial products by our competitors or us; |
|
·
| |
developments concerning proprietary rights, including patents; |
|
·
| |
developments concerning our collaborations; |
|
·
| |
publicity regarding actual or potential medical results relating to products under development by our competitors or us; |
|
·
| |
regulatory developments in the United States and foreign countries; |
|
·
| |
litigation or arbitration; |
|
·
| |
economic and other external factors or other disaster or crisis; and |
|
·
| |
period-to-period fluctuations in financial results. |
If we fail to continue to meet the listing standards of NASDAQ, our common stock may be delisted, which could have a material adverse effect on the liquidity of our common stock.
Our common stock is currently listed on the Nasdaq Global Market. The NASDAQ Stock Market LLC has requirements that a company must meet in order to remain listed on NASDAQ. In particular, NASDAQ rules require us to maintain a minimum bid price of $1.00 per share of our common stock. If the closing bid price of our common stock were to fall below $1.00 per share for 30 consecutive trading days or we do not meet other listing requirements, we would fail to be in compliance with NASDAQ’s listing standards. There can be no assurance that we will continue to meet the minimum bid price requirement, or any other requirement in the future. If we fail to meet the minimum bid price requirement, The NASDAQ Stock Market LLC may initiate the delisting process with a notification letter. If we were to receive such a notification, we would be afforded a grace period of 180 calendar days to regain compliance with the minimum bid price requirement. In order to regain compliance, shares of our common stock would need to maintain a minimum closing bid price of at least $1.00 per share for a minimum of 10 consecutive trading days. In addition, we may be unable to meet other applicable NASDAQ listing requirements, including maintaining minimum levels of stockholders’ equity or market values of our common stock in which case, our common stock could be delisted. If our common stock were to be delisted, the liquidity of our common stock would be adversely affected and the market price of our common stock could decrease.
Our ability to generate revenues will be diminished if we or our collaborative partners fail to obtain acceptable prices or an adequate level of reimbursement for products from third-party payers or government agencies.
The drugs we hope to develop may be rejected by the marketplace due to many factors, including cost. Our ability to commercially exploit a drug may be limited due to the continuing efforts of government and third-party payers to contain or reduce the costs of health care through various means. For example, in some foreign markets, pricing and profitability of prescription pharmaceuticals are subject to government control. In the United States, we expect that there will continue to be a number of federal and state proposals to implement similar government control. In addition, increasing emphasis on managed care in the United States will likely continue to put pressure on the pricing of pharmaceutical products. Cost control initiatives could decrease the price that we or any of our collaborators would receive for any products in the future. Further, cost control initiatives could adversely affect our and our collaborators’ ability to commercialize our products and our ability to realize royalties from this commercialization.
Our ability to commercialize pharmaceutical products with collaborators may depend, in part, on the extent to which reimbursement for the products will be available from:
|
·
| |
government and health administration authorities; |
|
·
| |
private health insurers; and |
|
·
| |
other third-party payers. |
Significant uncertainty exists as to the reimbursement status of newly-approved healthcare products. Third-party payers, including Medicare, are challenging the prices charged for medical products and services. Government and other third-party payers increasingly are attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for new drugs and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has not granted labeling approval. Third- party insurance coverage may not be available to patients for any products we discover and develop, alone or with collaborators. If government and other third-party payers do not provide adequate coverage and reimbursement levels for our products, the market acceptance of these products may be reduced.
If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit commercialization of our products.
The testing and marketing of medical products entail an inherent risk of product liability. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products. We carry product liability insurance that is limited in scope and amount and may not be adequate to fully protect us against product liability claims. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with corporate collaborators. We, or our corporate collaborators, might not be able to obtain insurance at a reasonable cost, if at all. While under various circumstances we are entitled to be indemnified against losses by our corporate collaborators, indemnification may not be available or adequate should any claim arise.
We depend on various scientific consultants and advisors for the success and continuation of our research and development efforts.
We work extensively with various scientific consultants and advisors. The potential success of our drug discovery and development programs depends, in part, on continued collaborations with certain of these consultants and advisors. We, and various members of our management and research staff, rely on certain of these consultants and advisors for expertise in our research, regulatory and clinical efforts. Our scientific advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. We do not know if we will be able to maintain such consulting agreements or that such scientific advisors will not enter into consulting arrangements, exclusive or otherwise, with competing pharmaceutical or biotechnology companies, any of which would have a detrimental impact on our research objectives and could have a material adverse effect on our business, financial condition and results of operations.
If we use biological and hazardous materials in a manner that causes injury or violates laws, we may be liable for damages, penalties or fines.
Our research and development activities involve the controlled use of potentially harmful biological materials as well as hazardous materials, chemicals and various radioactive compounds. We cannot completely eliminate the risk of accidental contamination or injury from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for damages that result or for penalties or fines that may be imposed, and such liability could exceed our resources. We are also subject to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with, or any potential violation of, these laws and regulations could be significant.
Our internal computer systems, or those used by our contract research organizations or other contractors or consultants, may fail or suffer security breaches.
Despite the implementation of security measures, our internal computer systems and those of our contract research organizations and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing clinical trials for a product candidate could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of any product candidates could be delayed.
Our facilities are located near known earthquake fault zones, and the occurrence of an earthquake or other catastrophic disaster could cause damage to our facilities and equipment, which could require us to cease or curtail operations.
Our facilities are located in the San Francisco Bay Area near known earthquake fault zones and are vulnerable to significant damage from earthquakes. We are also vulnerable to damage from other types of disasters, including fires, floods, power loss, communications failures and similar events. If any disaster were to occur, our ability to operate our business at our facilities would be seriously, or potentially completely, impaired, and our research could be lost or destroyed. In addition, the unique nature of our research activities and of much of our equipment could make it difficult for us to recover from a disaster. The insurance we maintain may not be adequate to cover our losses resulting from disasters or other business interruptions.
Future equity issuances or a sale of a substantial number of shares of our common stock may cause the price of our common stock to decline.
Because we will continue to need additional capital in the future to continue to expand our business and our research and development activities, among other things, we may conduct additional equity offerings. For example, under the universal shelf registration statement filed by us in May 2015 and declared effective by the SEC in July 2015, we may offer and sell any combination common stock, preferred stock, debt securities and warrants in one or more offerings, up to a cumulative value of $150 million. If we or our stockholders sell, or if it is perceived that we or they will sell, substantial amounts of our common stock (including pursuant to our Sales Agreement with Cantor or shares issued upon the exercise of options and warrants) in the public market, the market price of our common stock could fall. A decline in the market price of our common stock could make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem appropriate. In addition, future sales by us of our common stock, including pursuant to our Sales Agreement with Cantor, may be dilutive to existing stockholders. Furthermore, if we obtain funds through a credit facility or through the issuance of debt or preferred securities, these securities would likely have rights senior to the rights of our common stockholders, which could impair the value of our common stock.
Anti-takeover provisions in our charter documents and under Delaware law may make an acquisition of us, which may be beneficial to our stockholders, more difficult.
Provisions of our amended and restated certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us, even if doing so would benefit our stockholders. These provisions:
|
·
| |
establish that members of the board of directors may be removed only for cause upon the affirmative vote of stockholders owning a majority of our capital stock; |
|
·
| |
authorize the issuance of “blank check” preferred stock that could be issued by our board of directors to increase the number of outstanding shares and thwart a takeover attempt; |
|
·
| |
limit who may call a special meeting of stockholders; |
|
·
| |
prohibit stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; |
|
·
| |
establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings; |
|
·
| |
provide for a board of directors with staggered terms; and |
|
·
| |
provide that the authorized number of directors may be changed only by a resolution of our board of directors. |
In addition, Section 203 of the Delaware General Corporation Law, which imposes certain restrictions relating to transactions with major stockholders, may discourage, delay or prevent a third party from acquiring us.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
We currently lease facilities consisting of approximately 147,000 square feet of research and office space located at 1180 Veterans Boulevard, South San Francisco, California, of which, commencing in December 2014, we sublet approximately 57,000 square feet of our research and office space to an unrelated third party. Both the lease and the sublease expire in January 2018. We believe our facilities are in good operating condition and that the leased real property that we still occupy is adequate for all present and near term uses.
Item 3. Legal Proceedings
None.
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock commenced trading publicly under the symbol “RIGL” on December 7, 2000. The following table sets forth, for the periods indicated, the high and low intraday sales prices of our common stock as reported on the Nasdaq Global Market:
|
|
|
|
|
|
|
|
|
|
|
|
High
|
|
Low
|
|
|
Year Ended December 31, 2014
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
5.00
|
|
$
|
2.78
|
|
|
Second Quarter
|
|
$
|
4.20
|
|
$
|
2.64
|
|
|
Third Quarter
|
|
$
|
3.85
|
|
$
|
1.94
|
|
|
Fourth Quarter
|
|
$
|
2.57
|
|
$
|
1.56
|
|
|
Year Ended December 31, 2015
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
3.91
|
|
$
|
2.02
|
|
|
Second Quarter
|
|
$
|
5.20
|
|
$
|
2.95
|
|
|
Third Quarter
|
|
$
|
3.39
|
|
$
|
2.32
|
|
|
Fourth Quarter
|
|
$
|
3.68
|
|
$
|
2.42
|
|
On February 29, 2016, the last reported sale price for our common stock on the Nasdaq Global Market was $2.27 per share.
Holders
As of February 29, 2016, there were approximately 94 stockholders of record of our common stock.
Dividends
We have not paid any cash dividends on our common stock and currently do not plan to pay any cash dividends in the foreseeable future.
Performance Measurement Comparison
The graph below shows the cumulative total stockholder return of an investment of $100 (and the reinvestment of any dividends thereafter) on December 31, 2010 in (i) our common stock, (ii) the Nasdaq Composite Index and (iii) the Nasdaq Biotechnology Index. The Nasdaq Biotechnology Index is a modified‑capitalization weighted index that includes securities of Nasdaq‑listed companies classified according to the Industry Classification Benchmark as either Biotechnology or Pharmaceuticals and which also meet other eligibility criteria. Our stock price performance shown in the graph below is based upon historical data and is not indicative of future stock price performance.
The following graph and related information shall not be deemed “soliciting material” or be deemed to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing, except to the extent that we specifically incorporate it by reference into such filing.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Rigel Pharmaceuticals, Inc., the NASDAQ Composite Index
and the NASDAQ Biotechnology Index
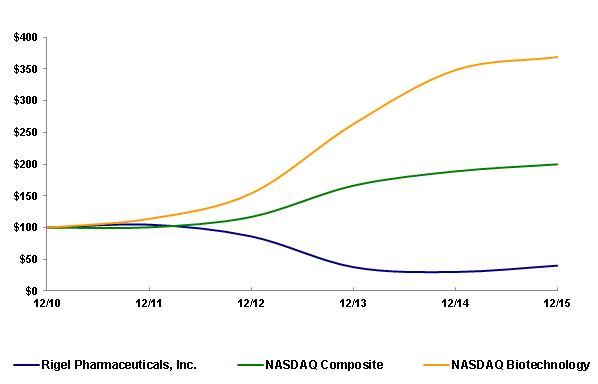
*$100 invested on December 31, 2010 in stock or index‑including reinvestment of dividends at fiscal year ending December 31.
Item 6. Selected Financial Data
The following selected financial data have been derived from our audited financial statements. The information set forth below is not necessarily indicative of our results of future operations and should be read in conjunction with “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Item 8. Financial Statements and Supplementary Data” included elsewhere in this Annual Report on Form 10‑ K.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fiscal Year Ended December 31,
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
2012
|
|
2011
|
|
|
|
|
(in thousands, except per share amounts)
|
|
|
Statements of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contract revenues from collaborations
|
|
$
|
28,895
|
|
$
|
8,250
|
|
$
|
7,150
|
|
$
|
2,250
|
|
$
|
4,750
|
|
|
Costs and expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
62,825
|
|
|
67,696
|
|
|
75,328
|
|
|
78,778
|
|
|
69,350
|
|
|
General and administrative
|
|
|
17,813
|
|
|
22,501
|
|
|
19,612
|
|
|
22,849
|
|
|
21,768
|
|
|
Loss on sublease
|
|
|
—
|
|
|
9,302
|
|
|
—
|
|
|
—
|
|
|
—
|
|
|
Restructuring charges
|
|
|
—
|
|
|
—
|
|
|
1,679
|
|
|
—
|
|
|
—
|
|
|
Total costs and expenses
|
|
|
80,638
|
|
|
99,499
|
|
|
96,619
|
|
|
101,627
|
|
|
91,118
|
|
|
Loss from operations
|
|
|
(51,743)
|
|
|
(91,249)
|
|
|
(89,469)
|
|
|
(99,377)
|
|
|
(86,368)
|
|
|
Interest income
|
|
|
222
|
|
|
243
|
|
|
426
|
|
|
520
|
|
|
420
|
|
|
Interest expense
|
|
|
—
|
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(25)
|
|
|
Gain on disposal of assets
|
|
|
57
|
|
|
98
|
|
|
16
|
|
|
17
|
|
|
—
|
|
|
Net loss
|
|
$
|
(51,464)
|
|
$
|
(90,908)
|
|
$
|
(89,027)
|
|
$
|
(98,840)
|
|
$
|
(85,973)
|
|
|
Net loss per share, basic and diluted
|
|
$
|
(0.58)
|
|
$
|
(1.04)
|
|
$
|
(1.02)
|
|
$
|
(1.32)
|
|
$
|
(1.36)
|
|
|
Weighted average shares used in computing net loss per share, basic and diluted
|
|
|
88,434
|
|
|
87,662
|
|
|
87,288
|
|
|
74,967
|
|
|
63,329
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
2012
|
|
2011
|
|
|
|
|
(in thousands)
|
|
|
Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and short-term investments
|
|
$
|
126,276
|
|
$
|
143,159
|
|
$
|
211,975
|
|
$
|
298,241
|
|
$
|
247,640
|
|
|
Working capital
|
|
|
95,228
|
|
|
136,512
|
|
|
209,781
|
|
|
290,254
|
|
|
238,706
|
|
|
Total assets
|
|
|
131,747
|
|
|
154,135
|
|
|
226,058
|
|
|
310,043
|
|
|
257,106
|
|
|
Accumulated deficit
|
|
|
(991,646)
|
|
|
(940,182)
|
|
|
(849,274)
|
|
|
(760,247)
|
|
|
(661,407)
|
|
|
Total stockholders’ equity
|
|
|
91,381
|
|
|
128,246
|
|
|
208,251
|
|
|
289,096
|
|
|
236,149
|
|
See Note 1 to the Financial Statements for description of the number of shares used in the computation of basic and diluted loss per share.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Overview
We are a clinical‑stage biotechnology company dedicated to the discovery and development of novel, targeted drugs in the therapeutic areas of immunology, oncology and immuno-oncology. Our pioneering research focuses on signaling pathways that are critical to disease mechanisms. Our current clinical programs include fostamatinib, an oral spleen tyrosine kinase (SYK) inhibitor, which is in Phase 3 clinical trials for immune thrombocytopenic purpura (ITP); a Phase 2 clinical trial for autoimmune hemolytic anemia (AIHA); and a Phase 2 clinical trial for IgA nephropathy (IgAN). In addition, we have two oncology product candidates in Phase 1 development with partners BerGenBio AS (BergenBio) and Daiichi Sankyo (Daiichi).
Since inception, we have financed our operations primarily through the sale of equity securities and contract payments under our collaboration agreements. Our research and development activities, including preclinical studies and clinical trials, consume substantial amounts of capital. As of December 31, 2015, we had approximately $126.3 million in cash, cash equivalents and short‑term investments. During the year ended December 31, 2015, we received an aggregate of $41.5 million upfront payments pursuant to our agreements with our collaborative partners. During the year ended December 31, 2015, approximately 1,722,312 shares of our common stock were sold under the Controlled Equity OfferingSM Sales Agreement with Cantor Fitzgerald & Co. (Cantor) Sales Agreement with aggregate net proceeds of $5.5 million. In December 2014, we entered into a sublease agreement with an unrelated third party to occupy a portion of our research and office space wherein we received approximately $3.9 million of sublease income and reimbursements in 2015. We believe that our existing capital resources will be sufficient to support our current and projected funding requirements into the third quarter of 2017. Unless and until we are able to generate a sufficient amount of product, royalty or milestone revenue, we expect to finance future cash needs through public and/or private offerings of equity securities, debt financings and/or collaboration and licensing arrangements.
Our revenues have consisted primarily of revenues from sponsored research and license agreements with our corporate collaborators. We earned contract revenues from collaborations of $28.9 million in 2015 comprised of the amortization of the $30.0 million upfront payment from BMS of $16.6 million and the FTE fees we earned from BMS of $822,000 as well as the upfront payments received from our other collaborative partners in the aggregate of $11.5 million. We expect our revenues to fluctuate from period to period and there can be no assurance that new collaborations will continue beyond their initial terms or that we will be able to receive any future contingent payments payable to us in the next twelve months or thereafter. Our potential future revenues may include payments from our current partners and from new partners with whom we enter into agreements in the future, if any, the timing and amount of which is unknown at this time.
Within our product development portfolio, our most advance program is fostamatinib in ITP. The first trial of our Phase 3 clinical program for ITP completed patient enrollment in January 2016. Our second Phase 3 trial is currently actively enrolling patients. We expect to separately report top line results of the two Phase 3 trials, with the first trial reporting in the middle of 2016 and the other trial reporting shortly thereafter. The results of preliminary or mid-stage studies do not necessarily predict clinical or commercial success, and large-stage clinical trials may fail to confirm the results observed in the previous studies. We can make no assurances regarding the likely results from our current or future clinical trials or the impact of those results on our business.
Product Development Programs
Our product development portfolio features multiple novel, targeted drug candidates in the therapeutic areas of immunology, oncology and immuno-oncology. Please refer to “Part I. Item 1. Business—Product Development Programs” for a detailed discussion of our multiple product candidates in development.
Corporate Collaborations
We conduct research and development programs independently and in connection with our corporate collaborators. Please refer to “Part I. Item 1. Business—Corporate Collaborations” for a detailed discussion of our corporate collaborations.
Research and Development Expenses
Our research and development expenditures include costs related to preclinical and clinical trials, scientific personnel, supplies, equipment, consultants, sponsored research, stock‑based compensation, and allocated facility costs.
We do not track fully‑burdened research and development costs separately for each of our drug candidates. We review our research and development expenses by focusing on three categories: research, development, and other. Our research team is focused on creating a portfolio of product candidates that can be developed into small‑molecule therapeutics in our own proprietary programs or with potential collaborative partners and utilizes our robust discovery engine to rapidly discover and validate new product candidates in our focused range of therapeutic indications. “Research” expenses relate primarily to personnel expenses, lab supplies, fees to third party research consultants and compounds. Our development group leads the implementation of our clinical and regulatory strategies and prioritizes disease indications in which our compounds may be studied in clinical trials. “Development” expenses relate primarily to clinical trials, personnel expenses, lab supplies and fees to third party research consultants. “Other” expenses primarily consist of allocated facilities costs and allocated stock‑based compensation expense relating to personnel in research and development groups.
In addition to reviewing the three categories of research and development expenses described in the preceding paragraph, we principally consider qualitative factors in making decisions regarding our research and development programs, which include enrollment in clinical trials and the results thereof, the clinical and commercial potential for our drug candidates and competitive dynamics. We also make our research and development decisions in the context of our overall business strategy, which includes the evaluation of potential collaborations for the development of our drug candidates.
The following table presents our total research and development expenses by category.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
From January 1, 2007*
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
to December 31, 2015
|
|
|
|
|
|
(in thousands)
|
|
|
Categories:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research
|
|
|
$
|
21,904
|
|
$
|
18,388
|
|
$
|
22,348
|
|
$
|
196,499
|
|
|
Development
|
|
|
|
25,988
|
|
|
27,727
|
|
|
31,915
|
|
|
283,282
|
|
|
Other
|
|
|
|
14,933
|
|
|
21,581
|
|
|
21,065
|
|
|
209,365
|
|
|
|
|
|
$
|
62,825
|
|
$
|
67,696
|
|
$
|
75,328
|
|
$
|
689,146
|
|
*We started tracking research and development expenses by category on January 1, 2007.
“Other” expenses mainly represent allocated facilities costs of approximately $10.8 million, $16.9 million and $17.1 million for the years ended December 31, 2015, 2014 and 2013, respectively, and allocated stock‑based compensation expenses of approximately $4.1 million, $4.7 million and $3.9 million for the years ended December 31, 2015, 2014 and 2013, respectively.
For the years ended December 31, 2015 and 2014, a major portion of our total research and development expense was associated with salaries of our research and development personnel, allocated facilities costs, and research and development expense for our ITP and IgAN programs. For the year ended December 31, 2013, a major portion of our total research and development expense was associated with salaries of our research and development personnel, allocated facilities costs, and our research and development expense for our asthma program, our topical JAK/SYK inhibitor program, as well as our oral SYK inhibitor program in ITP.
We do not have reliable estimates regarding the timing of our clinical trials. Preclinical testing and clinical development are long, expensive and uncertain processes. In general, biopharmaceutical development involves a series of steps, beginning with identification of a potential target and including, among others, proof of concept in animals and Phase 1, 2 and 3 clinical trials in humans. Significant delays in clinical testing could materially impact our product
development costs and timing of completion of the clinical trials. We do not know whether planned clinical trials will begin on time, will need to be halted or revamped or will be completed on schedule, or at all. Clinical trials can be delayed for a variety of reasons, including delays in obtaining regulatory approval to commence a trial, delays from scale up, delays in reaching agreement on acceptable clinical trial agreement terms with prospective clinical sites, delays in obtaining institutional review board approval to conduct a clinical trial at a prospective clinical site or delays in recruiting subjects to participate in a clinical trial.
We currently do not have reliable estimates of total costs for a particular drug candidate to reach the market. Our potential products are subject to a lengthy and uncertain regulatory process that may involve unanticipated additional clinical trials and may not result in receipt of the necessary regulatory approvals. Failure to receive the necessary regulatory approvals would prevent us from commercializing the product candidates affected. In addition, clinical trials of our potential products may fail to demonstrate safety and efficacy, which could prevent or significantly delay regulatory approval.
For further discussion on research and development activities, see “Research and Development Expense” under “Results of Operations” below.
Critical Accounting Policies and the Use of Estimates
Our discussion and analysis of our financial condition and results of operations is based upon our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles (U.S. GAAP). The preparation of these financial statements requires us to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. We evaluate our estimates, including those related to our stock‑based compensation, impairment issues, the estimated useful life of assets, estimated research term on our collaboration agreement with BMS, and estimated accruals, particularly research and development accruals, on an on‑going basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. We believe that there were no significant changes in our critical accounting policies during the year ended December 31, 2015 as compared to those previously disclosed in our Annual Report on Form 10‑K for the year ended December 31, 2014. We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our financial statements:
Revenue Recognition
We present revenue from our collaboration arrangements under the FASB ASC 808, Collaboration Arrangements. The terms of these agreements generally contain multiple elements, or deliverables, which may include (i) granting of license rights to our program, (ii) participation in a joint research committee, (iii) performance of research activities, and (iv) clinical supply and materials. The payments we receive under these arrangements typically include one or more of the following: non-refundable, up-front fees; funding of research and/or development efforts; contingent fees due upon the achievement of specified triggering events; and/or royalties on future product sales. We recognize revenue for the performance of services or the delivery of products when each of the following four criteria is met: (i) persuasive evidence of an arrangement exists; (ii) products are delivered or as services are rendered; (iii) the sales price is fixed or determinable; and (iv) collectability is reasonably assured.
Our revenue arrangements with multiple elements are evaluated under FASB ASC 605‑25, Multiple Element Arrangements, and are divided into separate units of accounting if certain criteria are met, including whether the deliverables have stand-alone value, based on the relevant facts and circumstances for each arrangement. The consideration we receive under collaboration arrangements is allocated among the separate units of accounting based on the selling price hierarchy, and the applicable revenue recognition criteria is applied to each of the separate units. We make significant judgments and estimates in the allocation of the consideration among the deliverables under the agreement, as well as the determination of the periods the units will be delivered to our collaborators. If there are deliverables in an arrangement that are not separable from other aspects of the contractual relationship, they are treated as a combined unit of accounting, with the allocated revenue for the combined unit recognized in a manner consistent
with the revenue recognition applicable to the final deliverable in the combined unit. Payments received prior to satisfying the relevant revenue recognition criteria are recorded as deferred revenue in the accompanying balance sheets and recognized as revenue when the revenue recognition criteria are met.
We typically receive non-refundable, up-front payments when licensing our intellectual property, which often occurs in conjunction with a research and development agreement. If we believe that the license to our intellectual property has stand-alone value, we generally recognize revenue attributed to the license upon delivery provided that there are no future performance requirements for use of the license. When we believe that the license to our intellectual property does not have stand-alone value, we would recognize revenue attributed to the license ratably from the effective date of the agreement or the delivery of the license up to the estimated completion date of the undelivered performance obligation. Revenues related to the research services with our corporate collaborators are recognized as research services are performed over the related research period. Under these agreements, we are required to perform research activities as specified in the agreement. The payments received are not refundable and are based on a contractual cost per full-time equivalent employee working on the project. Our research and development expenses under the collaborative research agreements approximate the revenue recognized under such agreements over the research period.
Revenues associated with substantive, at-risk milestones pursuant to collaborative agreements are recognized upon achievement of the milestones. We consider a milestone to be substantive at the inception of the arrangement if it is commensurate with either our performance to achieve the milestone or the enhancement of the value of the delivered item as a result of a specific outcome resulting from our performance to achieve the milestone, it relates solely to past performance and it is reasonable relative to all of the deliverables and payment terms within the arrangement. Non-refundable contingent future amounts receivable in connection with future events specified in collaboration agreements that are not considered milestones such as payments contingent solely upon the passage of time or the result of our collaborator's performance will be recognized as revenue when the recognition criteria discussed above are met.
Stock‑Based Compensation
We grant options to purchase our common stock to our officers, directors and all other employees and consultants under our stock option plans. Eligible employees can also purchase shares of our common stock at a price per share equal to the lesser of 85% of the fair market value on the first day of the offering period or 85% of the fair market value on the purchase date under our employee stock purchase plan (Purchase Plan). The benefits provided under these plans are stock‑based payments subject to the provisions of FASB ASC 718. We adopted the use of the straight‑line attribution method over the requisite service period for each entire stock award. In addition, we estimate the amount of expected forfeitures when calculating compensation costs, then record actual forfeitures as they occur. We review our forfeiture rates each quarter and make any necessary changes to our estimates.
The determination of the fair value of stock‑based payment awards on the date of grant using the Black‑Scholes option‑pricing model is affected by our stock price, as well as assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, volatility, expected term, risk‑free interest rate and dividends. We estimate volatility over the expected term of the option using historical share price performance. For expected term, among other things, we take into consideration our historical data of options exercised, cancelled and expired. The risk‑free rate is based on the U.S. Treasury constant maturity rate. We have not paid and do not expect to pay dividends in the foreseeable future. In order to calculate stock‑based compensation expense, we also estimate the forfeiture rate using our historical experience with options that cancel before they vest.
In December 2014, we entered into a severance agreement with our former CEO. As part of the severance arrangement we offered, we extended the date to which our former CEO had the right to exercise his vested options within 90 days from his termination date as was stipulated under his option agreement to the end of the contractual term of the options, of which the remaining contractual term for the most recently granted options is nine years.. In addition, we also accelerated the vesting period of certain of his unvested stock options. We determined the incremental fair value of the applicable options using the Black‑Scholes option‑pricing model based on assumptions that are subjective as discussed above. As a result of these modifications, we recorded incremental stock‑based compensation expense of approximately $1.5 million in the fourth quarter of 2014.
We granted options to purchase shares of common stock which will vest upon the achievement of certain performance-based milestones. We determined the fair values of these performance-based stock options using the Black-Scholes option pricing model at the date of grant. For the portion of the performance-based stock options of which the performance condition is considered probable of achievement, we recognized stock-based compensation expense on the related estimated fair value of such options on straight-line basis from the date of grant up to the date when we expect the performance condition will be achieved. For the performance conditions that are not considered probable of achievement at the grant date or upon quarterly re-evaluation, prior to the event actually occurring, we will recognize the related stock-based compensation expense when the event occurs or when we can determine that the performance condition is probable of achievement. In those cases, we will recognize the change in estimate at the time we determine the condition is probable of achievement (by recognizing stock-based compensation expense as cumulative catch-up adjustment as if we had estimated at the grant date that the performance condition would have been achieved) and recognize the remaining compensation cost up to the date when we expect the performance condition will be achieved, if any. During the year ended December 31, 2015, there were no changes in the estimated date of achievement in the performance condition related to the stock options granted in 2015 and 2014 that had material impact in our financial statements.
We also record charges associated with options granted to consultants reflecting the fair value and periodic fair value re‑measurement of outstanding consultant options under FASB ASC 505‑50. The valuation is based upon the current market value of our common stock and other assumptions, including the expected future volatility of our stock price, risk‑free interest rate and expected term. We amortize stock‑based compensation related to consultants using a straight‑line attribution method consistent with the method used for employees and with the attribution election we made upon adoption of FASB ASC 718.
Research and Development Accruals
We have various contracts with third parties related to our research and development activities. Costs that are incurred for services rendered, but not billed to us, as of the end of the period are estimated and accrued. We make estimates of the amounts incurred in each period based on the information available to us and our knowledge of the nature of the contractual activities generating such costs. Expenses related to other research and development contracts, such as research contracts, toxicology study contracts and manufacturing contracts are estimated to be incurred generally on a straight‑line basis over the duration of the contracts. Raw materials and study materials purchased for us by third parties are expensed at the time of purchase. Many of our estimates are based significantly or in part on information provided for us by third parties. If such information were not reported properly, our research and development expense amounts could be misstated.
Leases
We currently lease our research and office space under a noncancelable lease agreement with our landlord through January 2018. In December 2014, we entered into a sublease agreement with an unrelated third party to occupy a portion of our research and office space. In connection with this sublease, we recognized a loss on the sublease of $9.3 million during the fourth quarter of 2014. We record rent expense on a straight‑line basis for our lease, net of sublease income, wherein such arrangements contain scheduled rent increases over the term of the lease and sublease, respectively. For our sublease arrangement which we classified as an operating lease, our loss on the sublease is comprised of the present value of our future payments to our landlord less the present value of our future rent payments expected from our subtenant over the term of the sublease. The present value factor, which also affects the level of accreted interest expense that we will recognize as additional charges over the term of the lease, is based on our estimate of our credit‑risk adjusted borrowing rate at the time the initial sublease liability is calculated. Our estimate of our credit‑risk adjusted borrowing rate was based on our comparison of the rates used by other companies of our size, our financial condition at the time we entered into such sublease agreement, as well as other factors that would affect our credit worthiness.
Results of Operations
Year Ended December 31, 2015, 2014 and 2013
Revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Aggregate
|
|
Aggregate
|
|
|
|
|
|
Year Ended December 31,
|
|
Change
|
|
Change
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
2015 from 2014
|
|
2014 from 2013
|
|
|
|
|
|
(in thousands)
|
|
|
Contract revenues from collaborations
|
|
|
$
|
28,895
|
|
$
|
8,250
|
|
$
|
7,150
|
|
$
|
20,645
|
|
$
|
1,100
|
|
Contract revenues from collaborations of $28.9 million in 2015 were comprised of the amortization of the $30.0 million upfront payment from BMS of $16.6 million and the FTE fees we earned from BMS of $822,000 as well as the upfront payments received from our other collaborative partners in the aggregate of $11.5 million. Contract revenues from collaborations of $8.3 million in 2014 consisted of $5.8 million associated with the non‑refundable time‑based payment and $2.5 million payment from AZ for their continued development of R256 in asthma. Contract revenues from collaborations of $7.2 million in 2013 consisted of a $5.8 million payment associated with the non‑refundable time‑based payment from AZ resulting from AZ’s continued development of R256 in asthma, and a non‑refundable payment of $1.4 million from Daiichi related to an investigational new drug application filing for an oncology compound. As of December 31, 2015, deferred revenue related to the $30.0 million upfront payment from BMS was $13.4 million, which is expected to be recognized within revenue through September 2016. We had no deferred revenue as of December 31, 2014. Our potential future revenues may include payments from our current partners and from new partners with whom we enter into agreements in the future, if any, the timing and amount of which is unknown at this time.
Research and Development Expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Aggregate
|
|
Aggregate
|
|
|
|
|
|
Year Ended December 31,
|
|
Change
|
|
Change
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
2015 from 2014
|
|
2014 from 2013
|
|
|
|
|
|
(in thousands)
|
|
|
Research and development expense
|
|
|
$
|
62,825
|
|
$
|
67,696
|
|
$
|
75,328
|
|
$
|
(4,871)
|
|
$
|
(7,632)
|
|
|
Stock-based compensation expense included in research and development expense
|
|
|
$
|
4,100
|
|
$
|
4,674
|
|
$
|
3,930
|
|
$
|
(574)
|
|
$
|
744
|
|
The decrease in research and development expense for the year ended December 31, 2015, compared to the same period in 2014, was primarily due to the decrease in facilities costs resulting from the sublease agreement executed in December 2014, the completion in 2014 of a Phase 2 study of R348 in dry eye and the discontinuation of our indirect AMPK activator program, R118, in 2014, partially offset by the increases in bonus compensation expense and research and development costs related to our Phase 3 clinical program in ITP. The decrease in research and development expense for the year ended December 31, 2014, compared to the same period in 2013, was primarily due to the completion of two Phase 2 studies in 2013, completion of a Phase 2 study of R348 in dry eye in 2014, as well as the discontinuance of our indirect AMPK activator program, R118, in 2014. This was partially offset by the increase in research and development expense related to the fostamatinib in ITP and IgAN programs. We expect that our research and development expense will increase through 2016 due to the continued progress of our Phase 3 clinical trials in ITP and Phase 2 clinical trial in IgAN as well as our recently initiated Phase 2 clinical trial in AIHA in February 2016.
General and Administrative Expense
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Aggregate
|
|
Aggregate
|
|
|
|
|
|
|
Year Ended December 31,
|
|
Change
|
|
Change
|
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
2015 from 2014
|
|
2014 from 2013
|
|
|
|
|
|
|
(in thousands)
|
|
|
General and administrative expense
|
|
|
|
$
|
17,813
|
|
$
|
22,501
|
|
$
|
19,612
|
|
$
|
(4,688)
|
|
$
|
2,889
|
|
|
Stock-based compensation expense included in general and administrative expense
|
|
|
|
$
|
3,303
|
|
$
|
5,113
|
|
$
|
2,997
|
|
$
|
(1,810)
|
|
$
|
2,116
|
|
The decrease in general and administrative expense for the year ended December 31, 2015, compared to the same period in 2014, was primarily due to the decreases in personnel costs due to the retirement of our former Chief Executive Officer (CEO) in December 2014, as discussed below, and facilities costs as a result of the sublease agreement executed in December 2014. The increase in general and administrative expense for the year ended December 31, 2014, compared to the same period in 2013, was primarily due to the increase in stock‑based compensation expense and severance costs related to the retirement of our former CEO, partially offset by the increase in bonus compensation expense.
In December 2014, we entered into a severance agreement with our former CEO. The severance agreement provides for cash severance payments payable in installments over a duration of 18 months beginning on January 1, 2015. As part of the severance arrangement we offered, we extended the date to which our former CEO had the right to exercise his vested options within 90 days from his termination date as was stipulated under his option agreement to the end of the contractual term of the options, of which the remaining contractual term for the most recently granted options is nine years. In addition, we also accelerated the vesting period of certain of his unvested stock options. As a result of these modifications, we recorded incremental stock-based compensation expense in the fourth quarter of 2014.
Loss on Sublease
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended
|
|
Aggregate
|
|
Aggregate
|
|
|
|
|
December 31,
|
|
Change
|
|
Change
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
2015 from 2014
|
|
2014 from 2013
|
|
|
|
|
(in thousands)
|
|
|
Loss on sublease
|
|
$
|
—
|
|
$
|
9,302
|
|
$
|
—
|
|
$
|
(9,302)
|
|
$
|
9,302
|
|
In December 2014, we entered into a sublease arrangement whereby we sublet 56,750 square feet or approximately 39% of our research and office space and recorded a loss on sublease of $9.3 million. The loss on the sublease is derived from the present value of the excess of our future remaining payments to our landlord associated with the applicable subleased space over our contractual sublease income from our subtenant over the term of the sublease.
Restructuring Charges
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended
|
|
Aggregate
|
|
Aggregate
|
|
|
|
|
December 31,
|
|
Change
|
|
Change
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
2015 from 2014
|
|
2014 from 2013
|
|
|
|
|
(in thousands)
|
|
|
Restructuring charges
|
|
$
|
—
|
|
$
|
—
|
|
$
|
1,679
|
|
$
|
—
|
|
$
|
(1,679)
|
|
|
Stock-based compensation expense included in restructuring charges
|
|
$
|
—
|
|
$
|
—
|
|
$
|
239
|
|
$
|
—
|
|
$
|
(239)
|
|
In September 2013, we reduced our workforce by 30 positions, mostly from the drug discovery area as a consequence of prioritizing projects and efforts to conserve our working capital. We recorded restructuring charges of approximately $1.7 million, including $1.5 million of workforce reduction costs paid in cash, and $239,000 of non‑cash stock‑based compensation expense primarily as a result of the extension of the date to which the terminated employees had to exercise their vested options through June 30, 2014.
Interest income
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended
|
|
Aggregate
|
|
Aggregate
|
|
|
|
|
|
December 31,
|
|
Change
|
|
Change
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
2015 from 2014
|
|
2014 from 2013
|
|
|
|
|
|
(in thousands)
|
|
|
Interest income
|
|
|
$
|
222
|
|
$
|
243
|
|
$
|
426
|
|
$
|
(21)
|
|
$
|
(183)
|
|
Interest income results from our interest‑bearing cash and investment balances. The decrease in interest income for the year ended December 31, 2015, as compared to the same period in 2014, as well as for the year ended December 31, 2014, as compared to the same period in 2013, was primarily due to lower average cash balance of our short‑term investments.
Recent Accounting Pronouncements
In August 2014, the FASB issued ASU No. 2014‑15—Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern under ASC Subtopic 205‑40, Presentation of Financial Statements—Going Concern. ASU No. 2014‑15 provides guidance about management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. Management’s evaluation should be based on relevant conditions and events that are known and reasonably knowable at the date that the financial statements are issued (or at the date that the financial statements are available to be issued when applicable). Substantial doubt about an entity’s ability to continue as a going concern exists when relevant conditions and events, considered in the aggregate, indicate that it is probable that the entity will be unable to meet its obligations as they become due within one year after the date that the financial statements are issued (or available to be issued). ASU No. 2014‑15 is effective for the annual period ending after December 15, 2016 and early adoption is permitted. We will continue to evaluate the guidance under ASU No. 2014‑15 and present the required disclosures within our financial statements at the time of adoption. We plan to adopt this new standard in our annual financial statements for the year ending December 31, 2016 and we believe that the adoption of ASU No. 2014-15 will have no material effect on our financial statement disclosures.
In May 2014, the FASB issued ASU No. 2014-09—Revenue from Contracts with Customers, which supersedes the revenue recognition requirements under ASC Topic 605, Revenue Recognition, and most industry-specific guidance under the ASC. The core principle of ASU No. 2014-09 is that an entity should recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 defines a five step process to achieve this core principle and, in doing so, it is possible more judgment and estimates may be required within the revenue recognition process than required under existing U.S. GAAP including identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligation. ASU No. 2014-09 also requires additional disclosures to enable users of financial statements to understand the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts. ASU No. 2014-09 allows for either full retrospective or modified retrospective adoption, and we have not yet determined which approach we will apply. In July 2015, the FASB deferred by one year the effective date of ASU No. 2014-09 with the new effective date beginning after December 15, 2017, and the interim periods within that year and will allow early adoption for all entities as of the original effective date for public business entities, which was annual reporting periods beginning after December 15, 2016. We plan to adopt this new standard on January 1, 2018. We are currently evaluating the potential impact of the adoption of ASU No. 2014-09 on our financial statements and cannot estimate the impact of adoption at this time.
Liquidity and Capital Resources
Cash Requirements
From inception, we have financed our operations primarily through sales of equity securities, contract payments under our collaboration agreements and equipment financing arrangements. We have consumed substantial amounts of capital to date as we continue our research and development activities, including preclinical studies and clinical trials.
As of December 31, 2015, we had approximately $126.3 million in cash, cash equivalents and short‑term investments, as compared to approximately $143.2 million as of December 31, 2014, a decrease of approximately $16.9 million. The decrease was primarily attributable to the payments associated with funding our operating expenses for the year ended December 31, 2015, partially offset by the $41.5 million upfront payments received pursuant to our agreements with our collaborative partners. In August 2015, we entered into a Controlled Equity OfferingSM Sales Agreement with Cantor, as sales agent, pursuant to which we may sell, through Cantor, up to an aggregate of $30.0 million in shares of our common stock. The common stock is being sold at prevailing market prices at the time of the sale, and, as a result, prices may vary. During the year ended December 31, 2015, approximately 1,722,312 shares of our common stock were sold under the Sales Agreement with aggregate net proceeds of $5.5 million. At December 31, 2015, we had approximately $24.3 million of common stock registered for sale under the Sales Agreement. In December 2014, we entered into a sublease agreement with an unrelated third party to occupy a portion of our research and office space. We expect to receive over $5.0 million in future sublease income (excluding our subtenant’s share of facility’s operating expenses) over the remaining term of the sublease through January 2018. During the year ended December 31, 2015, we received approximately $3.9 million of sublease income and reimbursements. We believe that our existing capital resources will be sufficient to support our current and projected funding requirements into the third quarter of 2017. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development of our product candidates and other research and development activities, including risks and uncertainties that could impact the rate of progress of our development activities, we are unable to estimate with certainty the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical trials and other research and development activities.
Our operations will require significant additional funding for the foreseeable future. Unless and until we are able to generate a sufficient amount of product, royalty or milestone revenue, we expect to finance future cash needs through public and/or private offerings of equity securities, debt financings and/or collaboration and licensing arrangements, and to a much lesser extent through interest income earned on the investment of our excess cash balances and short‑term investments. With the exception of contingent and royalty payments that we may receive under our existing collaborations, we do not currently have any committed future funding. To the extent we raise additional capital by issuing equity securities, our stockholders could at that time experience substantial dilution. Any debt financing that we are able to obtain may involve operating covenants that restrict our business. To the extent that we raise additional funds through collaboration and licensing arrangements, we may be required to relinquish some of our rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us.
Our future funding requirements will depend upon many factors, including, but not limited to:
|
·
| |
the progress and success of our clinical trials and preclinical activities (including studies and manufacture of materials) of our product candidates conducted by us; |
|
·
| |
the success of our corporate collaborations or license agreements; |
|
·
| |
the progress of research programs carried out by us; |
|
·
| |
any changes in the breadth of our research and development programs; |
|
·
| |
the ability to achieve the events identified in our collaborative agreements that trigger payments to us from our collaboration partners; |
|
·
| |
the progress of the research and development efforts of our collaborative partners; |
|
·
| |
our ability to manage our growth; |
|
·
| |
competing technological and market developments; |
|
·
| |
the costs and timing of obtaining, enforcing and defending our patent and other intellectual property rights; and |
|
·
| |
the costs and timing of regulatory filings and approvals by us and our collaborators. |
Insufficient funds may require us to delay, scale back or eliminate some or all of our research or development programs, to lose rights under existing licenses or to relinquish greater or all rights to product candidates at an earlier stage of development or on less favorable terms than we would otherwise choose or may adversely affect our ability to operate as a going concern.
For the years ended December 31, 2015 and 2014, we maintained an investment portfolio primarily in money market funds, U. S. treasury bills, government‑sponsored enterprise securities, and corporate bonds and commercial paper. Cash in excess of immediate requirements is invested with regard to liquidity and capital preservation. Wherever possible, we seek to minimize the potential effects of concentration and degrees of risk. We will continue to monitor the impact of the changes in the conditions of the credit and financial markets to our investment portfolio and assess if future changes in our investment strategy are necessary.
Cash Flows from Operating, Investing and Financing Activities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
|
|
(in thousands)
|
|
|
Net cash provided by (used in):
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities
|
|
$
|
(23,413)
|
|
$
|
(69,753)
|
|
$
|
(86,077)
|
|
|
Investing activities
|
|
|
44,613
|
|
|
62,932
|
|
|
72,396
|
|
|
Financing activities
|
|
|
7,053
|
|
|
1,170
|
|
|
1,051
|
|
|
Net increase (decrease) in cash and cash equivalents
|
|
$
|
28,253
|
|
$
|
(5,651)
|
|
$
|
(12,630)
|
|
Net cash used in operating activities was approximately $23.4 million in 2015 compared to approximately $69.8 million and $86.1 million in 2014 and 2013, respectively. Net cash used in operating activities primarily consisted of cash payments related to our research and development programs, partially offset by the $41.5 million upfront payments received pursuant to our agreements with our collaborative partners. The timing of cash requirements may vary from period to period depending on our research and development activities, including our planned preclinical and clinical trials, and future requirements to establish commercial capabilities for any products that we may develop.
Net cash provided by investing activities was approximately $44.6 million in 2015 compared to approximately $62.9 million and $72.4 million in 2014 and 2013, respectively. Net cash provided by investing activities in 2015, 2014 and 2013 related to net maturities of short‑term investments, partially offset by capital expenditures. Capital expenditures were approximately $546,000, $413,000 and $1.2 million in 2015, 2014 and 2013, respectively.
Net cash provided by financing activities was approximately $7.1 million in 2015 compared to approximately $1.2 million and $1.1 million in 2014 and 2013, respectively. Net cash provided by financing activities in 2015 consisted of net proceeds from issuance of shares under the Controlled Equity Offering Sales Agreement as well as proceeds from exercise of outstanding options and issuance of shares under the Purchase Plan. Net cash provided by financing activities in 2014 and 2013 related to the proceeds from the exercise of outstanding options and issuance of shares under the Purchase Plan.
Off‑Balance Sheet Arrangements
As of December 31, 2015, we had no off‑balance sheet arrangements (as defined in Item 303(a)(4)(ii) of Regulation S‑K under the Exchange Act).
Contractual Obligations
We conduct our research and development programs internally and through third parties that include, among others, arrangements with universities, consultants and contract research organizations. We have contractual arrangements with these parties, however our contracts with them are cancelable generally on reasonable notice within one year and our obligations under these contracts are primarily based on services performed. We do not have any purchase commitments under any collaboration arrangements.
As of December 31, 2015, we had the following contractual commitments:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Less than
|
|
Payment Due By Period
|
|
More than
|
|
|
|
|
Total
|
|
1 Year
|
|
1 - 3 Years
|
|
3 - 5 Years
|
|
5 Years
|
|
|
|
|
(in thousands)
|
|
|
Facilities lease(1)
|
|
$
|
33,034
|
|
$
|
15,530
|
|
$
|
17,504
|
|
$
|
—
|
|
$
|
—
|
|
|
(1)
| |
In December 2014, we entered into a sublease agreement with an unrelated third party to lease up a portion of the research and office space. The facilities lease obligations above do not include the sublease income of $5.8 million over the remaining term of the sublease. |
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. Some of the securities in which we invest may have market risk. This means that a change in prevailing interest rates may cause the fair value amount of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then‑prevailing rate and the prevailing interest rate later rises, the market value amount of our investment will decline. To minimize this risk, we maintain our portfolio of cash equivalents and short‑term investments in a variety of securities, including money market funds and government and non‑government debt securities and the maturities of each of these instruments is less than one year. In 2015, we maintained an investment portfolio primarily in money market funds, U. S. treasury bills, government‑sponsored enterprise securities, and corporate bonds and commercial paper. Due to the primarily short‑term nature and low interest rate yields of these investments, we believe we do not have a material exposure to interest rate risk and market risk arising from our investments. Therefore, no quantitative tabular disclosure is provided.
We have operated primarily in the United States, and all funding activities with our contract research organizations to date have been made in U.S. dollars. Accordingly, we have not had any significant exposure to foreign currency rate fluctuations.
Item 8. Financial Statements and Supplementary Data
INDEX TO FINANCIAL STATEMENTS
Rigel Pharmaceuticals, Inc.
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Rigel Pharmaceuticals, Inc.
We have audited the accompanying balance sheets of Rigel Pharmaceuticals, Inc. as of December 31, 2015 and 2014, and the related statements of operations, comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2015. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Rigel Pharmaceuticals, Inc. at December 31, 2015 and 2014, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2015, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Rigel Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 8, 2016 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Redwood City, California
March 8, 2016
RIGEL PHARMACEUTICALS, INC.
BALANCE SHEETS
(In thousands, except share and per share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2015
|
|
2014
|
|
|
|
|
|
|
|
|
|
|
|
Assets
|
|
|
|
|
|
|
|
|
Current assets:
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
43,456
|
|
$
|
15,203
|
|
|
Short-term investments
|
|
|
82,820
|
|
|
127,956
|
|
|
Accounts receivable
|
|
|
203
|
|
|
5,750
|
|
|
Prepaid and other current assets
|
|
|
2,545
|
|
|
1,628
|
|
|
Total current assets
|
|
|
129,024
|
|
|
150,537
|
|
|
Property and equipment, net
|
|
|
1,613
|
|
|
2,509
|
|
|
Other assets
|
|
|
1,110
|
|
|
1,089
|
|
|
|
|
$
|
131,747
|
|
$
|
154,135
|
|
|
Liabilities and stockholders’ equity
|
|
|
|
|
|
|
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
2,763
|
|
$
|
1,613
|
|
|
Accrued compensation
|
|
|
6,251
|
|
|
2,832
|
|
|
Accrued research and development
|
|
|
4,953
|
|
|
3,993
|
|
|
Other accrued liabilities
|
|
|
1,133
|
|
|
534
|
|
|
Deferred revenue
|
|
|
13,427
|
|
|
—
|
|
|
Deferred liability – sublease, current portion
|
|
|
3,005
|
|
|
2,803
|
|
|
Deferred rent, current portion
|
|
|
2,264
|
|
|
2,250
|
|
|
Total current liabilities
|
|
|
33,796
|
|
|
14,025
|
|
|
|
|
|
|
|
|
|
|
|
Long-term portion of deferred liability – sublease
|
|
|
3,460
|
|
|
6,466
|
|
|
Long-term portion of deferred rent
|
|
|
3,083
|
|
|
5,347
|
|
|
Other long-term liabilities
|
|
|
27
|
|
|
51
|
|
|
|
|
|
|
|
|
|
|
|
Commitments
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
Preferred stock, $0.001 par value; 10,000,000 shares authorized; none issued and outstanding as of December 31, 2015 and 2014
|
|
|
—
|
|
|
—
|
|
|
Common stock, $0.001 par value; 200,000,000 shares authorized; 90,554,589 and 88,041,445 shares issued and outstanding as of December 31, 2015 and 2014, respectively
|
|
|
91
|
|
|
88
|
|
|
Additional paid-in capital
|
|
|
1,082,980
|
|
|
1,068,347
|
|
|
Accumulated other comprehensive loss
|
|
|
(44)
|
|
|
(7)
|
|
|
Accumulated deficit
|
|
|
(991,646)
|
|
|
(940,182)
|
|
|
Total stockholders’ equity
|
|
|
91,381
|
|
|
128,246
|
|
|
|
|
$
|
131,747
|
|
$
|
154,135
|
|
See accompanying notes.
RIGEL PHARMACEUTICALS, INC.
STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
Contract revenues from collaborations
|
|
|
$
|
28,895
|
|
$
|
8,250
|
|
$
|
7,150
|
|
|
Costs and expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
|
62,825
|
|
|
67,696
|
|
|
75,328
|
|
|
General and administrative
|
|
|
|
17,813
|
|
|
22,501
|
|
|
19,612
|
|
|
Loss on sublease
|
|
|
|
—
|
|
|
9,302
|
|
|
—
|
|
|
Restructuring charges
|
|
|
|
—
|
|
|
—
|
|
|
1,679
|
|
|
Total costs and expenses
|
|
|
|
80,638
|
|
|
99,499
|
|
|
96,619
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
|
(51,743)
|
|
|
(91,249)
|
|
|
(89,469)
|
|
|
Interest income
|
|
|
|
222
|
|
|
243
|
|
|
426
|
|
|
Gain on disposal of assets
|
|
|
|
57
|
|
|
98
|
|
|
16
|
|
|
Net loss
|
|
|
$
|
(51,464)
|
|
$
|
(90,908)
|
|
$
|
(89,027)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share, basic and diluted
|
|
|
$
|
(0.58)
|
|
$
|
(1.04)
|
|
$
|
(1.02)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares used in computing net loss per share, basic and diluted
|
|
|
|
88,434
|
|
|
87,662
|
|
|
87,288
|
|
See accompanying notes.
RIGEL PHARMACEUTICALS, INC.
STATEMENTS OF COMPREHENSIVE LOSS
(In thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
Net loss
|
|
|
$
|
(51,464)
|
|
$
|
(90,908)
|
|
$
|
(89,027)
|
|
|
Other comprehensive loss:
|
|
|
|
|
|
|
|
|
|
|
|
|
Net unrealized loss on short-term investments
|
|
|
|
(37)
|
|
|
(54)
|
|
|
(35)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
$
|
(51,501)
|
|
$
|
(90,962)
|
|
$
|
(89,062)
|
|
See accompanying notes.
RIGEL PHARMACEUTICALS, INC.
STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except share and per share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
Other
|
|
|
|
|
Total
|
|
|
|
|
Common Stock
|
|
Paid-in
|
|
Comprehensive
|
|
Accumulated
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
Amount
|
|
Capital
|
|
Income (Loss)
|
|
Deficit
|
|
Equity
|
|
|
Balance at January 1, 2013
|
|
87,140,632
|
|
|
87
|
|
|
1,049,174
|
|
|
82
|
|
|
(760,247)
|
|
|
289,096
|
|
|
Net loss
|
|
—
|
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(89,027)
|
|
|
(89,027)
|
|
|
Net change in unrealized gain on short-term investments
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(35)
|
|
|
—
|
|
|
(35)
|
|
|
Issuance of common stock upon exercise of options and participation in Purchase Plan
|
|
383,717
|
|
|
1
|
|
|
1,050
|
|
|
—
|
|
|
—
|
|
|
1,051
|
|
|
Stock compensation expense
|
|
—
|
|
|
—
|
|
|
7,166
|
|
|
—
|
|
|
—
|
|
|
7,166
|
|
|
Balance at December 31, 2013
|
|
87,524,349
|
|
$
|
88
|
|
$
|
1,057,390
|
|
$
|
47
|
|
$
|
(849,274)
|
|
$
|
208,251
|
|
|
Net loss
|
|
—
|
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(90,908)
|
|
|
(90,908)
|
|
|
Net change in unrealized gain on short-term investments
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(54)
|
|
|
—
|
|
|
(54)
|
|
|
Issuance of common stock upon exercise of options and participation in Purchase Plan
|
|
517,096
|
|
|
—
|
|
|
1,170
|
|
|
—
|
|
|
—
|
|
|
1,170
|
|
|
Stock compensation expense
|
|
—
|
|
|
—
|
|
|
9,787
|
|
|
—
|
|
|
—
|
|
|
9,787
|
|
|
Balance at December 31, 2014
|
|
88,041,445
|
|
|
88
|
|
|
1,068,347
|
|
|
(7)
|
|
|
(940,182)
|
|
|
128,246
|
|
|
Net loss
|
|
—
|
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(51,464)
|
|
|
(51,464)
|
|
|
Net change in unrealized gain on short-term investments
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(37)
|
|
|
—
|
|
|
(37)
|
|
|
Issuance of common stock upon exercise of options and participation in Purchase Plan
|
|
790,832
|
|
|
1
|
|
|
1,760
|
|
|
—
|
|
|
—
|
|
|
1,761
|
|
|
Issuance of common stock, net of offering costs
|
|
1,722,312
|
|
|
2
|
|
|
5,470
|
|
|
—
|
|
|
—
|
|
|
5,472
|
|
|
Stock compensation expense
|
|
—
|
|
|
—
|
|
|
7,403
|
|
|
—
|
|
|
—
|
|
|
7,403
|
|
|
Balance at December 31, 2015
|
|
90,554,589
|
|
$
|
91
|
|
$
|
1,082,980
|
|
$
|
(44)
|
|
$
|
(991,646)
|
|
$
|
91,381
|
|
See accompanying notes.
RIGEL PHARMACEUTICALS, INC.
STATEMENTS OF CASH FLOWS
(In thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
Operating activities
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(51,464)
|
|
$
|
(90,908)
|
|
$
|
(89,027)
|
|
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization
|
|
|
1,439
|
|
|
2,359
|
|
|
2,592
|
|
|
Stock-based compensation expense
|
|
|
7,403
|
|
|
9,787
|
|
|
7,166
|
|
|
Gain on disposal of assets
|
|
|
(57)
|
|
|
(98)
|
|
|
(16)
|
|
|
Loss on sublease
|
|
|
—
|
|
|
9,302
|
|
|
—
|
|
|
Changes in assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable
|
|
|
5,547
|
|
|
—
|
|
|
(5,750)
|
|
|
Prepaid and other current assets
|
|
|
(917)
|
|
|
722
|
|
|
1,867
|
|
|
Other assets
|
|
|
159
|
|
|
439
|
|
|
231
|
|
|
Accounts payable
|
|
|
1,150
|
|
|
(2,290)
|
|
|
2,206
|
|
|
Accrued compensation
|
|
|
3,419
|
|
|
(17)
|
|
|
(3,926)
|
|
|
Accrued research and development
|
|
|
960
|
|
|
2,405
|
|
|
(536)
|
|
|
Other accrued liabilities
|
|
|
599
|
|
|
(212)
|
|
|
(196)
|
|
|
Deferred revenue
|
|
|
13,427
|
|
|
—
|
|
|
—
|
|
|
Deferred rent and other long term liabilities
|
|
|
(5,078)
|
|
|
(1,242)
|
|
|
(688)
|
|
|
Net cash used in operating activities
|
|
|
(23,413)
|
|
|
(69,753)
|
|
|
(86,077)
|
|
|
Investing activities
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of short-term investments
|
|
|
(151,763)
|
|
|
(218,594)
|
|
|
(308,846)
|
|
|
Maturities of short-term investments
|
|
|
196,862
|
|
|
281,705
|
|
|
365,968
|
|
|
Sales of short-term investments
|
|
|
—
|
|
|
—
|
|
|
16,479
|
|
|
Proceeds from disposal of assets
|
|
|
60
|
|
|
234
|
|
|
16
|
|
|
Capital expenditures
|
|
|
(546)
|
|
|
(413)
|
|
|
(1,221)
|
|
|
Net cash provided by investing activities
|
|
|
44,613
|
|
|
62,932
|
|
|
72,396
|
|
|
Financing activities
|
|
|
|
|
|
|
|
|
|
|
|
Net proceeds from issuances of common stock upon exercise of options and participation in Purchase Plan
|
|
|
1,761
|
|
|
1,170
|
|
|
1,051
|
|
|
Proceeds from issuance of common stock, net of offering costs
|
|
|
5,292
|
|
|
—
|
|
|
—
|
|
|
Net cash provided by financing activities
|
|
|
7,053
|
|
|
1,170
|
|
|
1,051
|
|
|
Net increase (decrease) in cash and cash equivalents
|
|
|
28,253
|
|
|
(5,651)
|
|
|
(12,630)
|
|
|
Cash and cash equivalents at beginning of period
|
|
|
15,203
|
|
|
20,854
|
|
|
33,484
|
|
|
Cash and cash equivalents at end of period
|
|
$
|
43,456
|
|
$
|
15,203
|
|
$
|
20,854
|
|
See accompanying notes.
In this Annual Report on Form 10‑K, “Rigel,” “we,” “us” and “our” refer to Rigel Pharmaceuticals, Inc. and “common stock” refers to Rigel’s common stock, par value $0.001 per share.
1. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Nature of operations and basis of presentation
We were incorporated in the state of Delaware on June 14, 1996. We are engaged in the discovery and development of novel, targeted drug candidates in the therapeutic areas of immunology, oncology and immuno-oncology.
Use of estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Significant estimates and assumptions made by management include those relating to our sublease agreement including the determination of discount rate used, stock-based compensation, impairment issues, estimated useful life of assets, estimated research term on our collaboration agreement with BMS, and estimated accruals, particularly research and development accruals. We believe that the estimates and judgments upon which we rely are reasonable based upon information available to us at the time that these estimates and judgments are made, however actual results could differ from these estimates. To the extent there are material differences between these estimates and actual results, our financial statements will be affected.
Stock award plans
We have three stock option plans, our 2011 Equity Incentive Plan (2011 Plan), 2000 Equity Incentive Plan (2000 Plan) and 2000 Non‑Employee Directors Stock Option Plan (Directors’ Plan), that provide for granting to our officers, directors and all other employees and consultants options to purchase shares of our common stock. We also have our Employee Stock Purchase Plan (Purchase Plan), where eligible employees can purchase shares of our common stock at a price per share equal to the lesser of 85% of the fair market value on the first day of the offering period or 85% of the fair market value on the purchase date. The fair value of each option award is estimated on the date of grant using the Black‑Scholes option pricing model which considered our stock price, as well as assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, volatility, expected term, risk‑free interest rate and dividends. We estimate volatility over the expected term of the option using historical share price performance. For expected term, we take into consideration our historical data of options exercised, cancelled and expired. The risk‑free rate is based on the U.S. Treasury constant maturity rate. We have not paid and do not expect to pay dividends in the foreseeable future. In order to calculate stock‑ based compensation expense, we also estimate the forfeiture rate using our historical experience with options that cancel before they vest. We review our forfeiture rates each quarter and make any necessary changes to our estimates. We use the straight‑line attribution method over the requisite employee service period for the entire award in recognizing stock‑based compensation expense.
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
We granted performance-based stock options to purchase shares of our common stock which will vest upon the achievement of certain corporate performance-based milestones. We determined the fair values of these performance-based stock options using the Black-Scholes option pricing model at the date of grant. For the portion of the performance-based stock options of which the performance condition is considered probable of achievement, we recognized stock-based compensation expense on the related estimated fair value of such options on a straight-line basis from the date of grant up to the date when we expect the performance condition will be achieved. For the performance conditions that are not considered probable of achievement at the grant date or upon quarterly re-evaluation, prior to the event actually occurring, we will recognize the related stock-based compensation expense when the event occurs or when we can determine that the performance condition is probable of achievement. In those cases, we will recognize the change in estimate at the time we determine the condition is probable of achievement (by recognizing stock-based compensation expense as cumulative catch-up adjustment as if we had estimated at the grant date that the performance condition would have been achieved) and recognize the remaining compensation cost up to the date when we expect the performance condition will be achieved, if any.
Cash, cash equivalents and short-term investments
We consider all highly liquid investments in debt securities with maturity from the date of purchase of 90 days or less to be cash equivalents. Cash equivalents consist of money market funds, U.S. treasury bills, corporate bonds and commercial paper and investments in government‑sponsored enterprises. Our short-term investments include U.S. treasury bills, obligations of government‑ sponsored enterprises and corporate bonds and commercial paper. By policy, we limit the concentration of credit risk by diversifying our investments among a variety of high credit‑quality issuers.
All cash equivalents and short‑term investments are classified as available‑for‑sale securities. Available‑for‑sale securities are carried at fair value at December 31, 2015 and 2014. Unrealized gains (losses) are reported in the statements of stockholders’ equity and comprehensive loss. Fair value is estimated based on available market information or valuation methodologies. The cost of securities sold is based on the specific identification method. See Note 5 for a summary of available-for-sale securities at December 31, 2015 and 2014.
Fair value of financial instruments
The carrying values of cash, accounts receivable, accounts payable and accrued liabilities approximate fair value due to the short-term maturity of those instruments. Cash equivalents and short-term investments are carried at fair value at December 31, 2015 and 2014.
Concentration of credit risk
Financial instruments that potentially subject us to concentrations of credit risk are primarily cash and cash equivalents, short-term investments and accounts receivable. Cash equivalents and short-term investments primarily consist of money market funds, U. S. treasury bills, government-sponsored enterprise securities, and corporate bonds and commercial paper. Due to the short-term nature of these investments, we believe we do not have a material exposure to credit risk arising from our investments. All cash and cash equivalents and short-term investments are maintained with financial institutions that management believes are creditworthy. As of December 31, 2015 and 2014, our accounts receivable primarily consisted of $203,000 from BMS relating to the performance of research activities and $5.8 million time-based non-refundable fee from AZ, respectively. To date, we have not experienced significant losses with respect to the collection of our accounts receivable and we believe that we do not have a material exposure to credit risk arising from our accounts receivable.
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
Property and equipment
Property and equipment are stated at cost. Depreciation is calculated using the straight‑line method over the estimated useful lives of the assets, which range from three to seven years.
Revenue recognition
We present revenue from our collaboration arrangements under the FASB ASC 808, Collaboration Arrangements. The terms of these agreements generally contain multiple elements, or deliverables, which may include (i) granting of license rights to our program, (ii) participation in a joint research committee, (iii) performance of research activities, and (iv) clinical supply and materials. The payments we receive under these arrangements typically include one or more of the following: non-refundable, up-front fees; funding of research and/or development efforts; contingent fees due upon the achievement of specified triggering events; and/or royalties on future product sales. We recognize revenue for the performance of services or the delivery of products when each of the following four criteria is met: (i) persuasive evidence of an arrangement exists; (ii) products are delivered or as services are rendered; (iii) the sales price is fixed or determinable; and (iv) collectability is reasonably assured.
Our revenue arrangements with multiple elements are evaluated under FASB ASC 605‑25, Multiple Element Arrangements, and are divided into separate units of accounting if certain criteria are met, including whether the deliverables have stand-alone value, based on the relevant facts and circumstances for each arrangement. The consideration we receive under collaboration arrangements is allocated among the separate units of accounting based on the selling price hierarchy, and the applicable revenue recognition criteria is applied to each of the separate units. We make significant judgments and estimates in the allocation of the consideration among the deliverables under the agreement, as well as the determination of the periods the units will be delivered to our collaborators. If there are deliverables in an arrangement that are not separable from other aspects of the contractual relationship, they are treated as a combined unit of accounting, with the allocated revenue for the combined unit recognized in a manner consistent with the revenue recognition applicable to the final deliverable in the combined unit. Payments received prior to satisfying the relevant revenue recognition criteria are recorded as deferred revenue in the accompanying balance sheets and recognized as revenue when the revenue recognition criteria are met.
We typically receive non-refundable, up-front payments when licensing our intellectual property, which often occurs in conjunction with a research and development agreement. If we believe that the license to our intellectual property has stand-alone value, we generally recognize revenue attributed to the license upon delivery provided that there are no future performance requirements for use of the license. When we believe that the license to our intellectual property does not have stand-alone value, we would recognize revenue attributed to the license ratably from the effective date of the agreement or the delivery of the license up to the estimated completion date of the undelivered performance obligation. Revenues related to the research services with our corporate collaborators are recognized as research services are performed over the related research period. Under these agreements, we are required to perform research activities as specified in the agreement. The payments received are not refundable and are based on a contractual cost per full-time equivalent employee working on the project. Our research and development expenses under the collaborative research agreements approximate the revenue recognized under such agreements over the research period.
Revenues associated with substantive, at-risk milestones pursuant to collaborative agreements are recognized upon achievement of the milestones. We consider a milestone to be substantive at the inception of the arrangement if it is commensurate with either our performance to achieve the milestone or the enhancement of the value of the delivered item as a result of a specific outcome resulting from our performance to achieve the milestone, it relates solely to past performance and it is reasonable relative to all of the deliverables and payment terms within the arrangement. Non-refundable contingent future amounts receivable in connection with future events specified in collaboration agreements that are not considered milestones such as payments contingent solely upon the passage of time or the result of our collaborator's performance will be recognized as revenue when the recognition criteria discussed above are met.
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
Research and development expenses
Research and development expenses include costs for scientific personnel, supplies, equipment, consultants, research sponsored by us, allocated facility costs, costs related to pre‑clinical and clinical trials, including raw materials, and stock‑based compensation expense. All such costs are charged to research and development expense as incurred and at the time raw materials are purchased.
Research and development accruals
We have various contracts with third parties related to our research and development activities. Costs that are incurred for services rendered, but not billed to us, as of the end of the period are estimated and accrued. We make estimates of the amounts incurred in each period based on the information available to us and our knowledge of the nature of the contractual activities generating such costs. Expenses related to other research and development contracts, such as research contracts, toxicology study contracts and manufacturing contracts are estimated to be incurred generally on a straight‑line basis over the duration of the contracts. Raw materials and study materials purchased for us by third parties are expensed at the time of purchase.
Leases
We currently lease our research and office space under a noncancelable lease agreement with our landlord through January 2018. In December 2014, we entered into a sublease agreement with an unrelated third party to occupy a portion of our research and office space. In connection with this sublease, we recognized a loss on the sublease of $9.3 million during the fourth quarter of 2014. We record rent expense on a straight‑line basis for our lease, net of sublease income, wherein such arrangements contain scheduled rent increases over the term of the lease and sublease, respectively. For our sublease arrangement which we classified as an operating lease, our loss on the sublease is comprised of the present value of our future payments to our landlord less the present value of our future rent payments expected from our subtenant over the term of the sublease. The present value factor, which also affects the level of accreted interest expense that we will recognize as additional charges over the term of the lease, is based on our estimate of our credit‑risk adjusted borrowing rate at the time the initial sublease liability is calculated. Our estimate of our credit‑risk adjusted borrowing rate was based on our comparison of the rates used by other companies of our size, our financial condition at the time we entered into such sublease agreement, as well as other factors that would affect our credit worthiness.
Contingencies
We are subject to claims related to the patent protection of certain of our technologies, as well as a purported securities class action lawsuit and other litigation. We are required to assess the likelihood of any adverse judgments or outcomes to these matters as well as potential ranges of probable losses. A determination of the amount of reserves required, if any, for these contingencies is made after careful analysis of each individual issue.
Income taxes
We use the asset and liability method to account for income taxes. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities from a change in tax rates is recognized in income in the period the change is enacted. A valuation allowance is established to reduce deferred tax assets to an amount whose realization is more likely than not.
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
Net loss per share
Basic net loss per share is computed by dividing net loss by the weighted‑average number of shares of common stock outstanding during the period. Diluted net loss per share is computed by dividing net loss by the weighted‑average number of shares of common stock outstanding during the period and the number of additional shares of common stock that would have been outstanding if potentially dilutive securities had been issued. Potentially dilutive securities include warrant and stock options and shares issuable under our Purchase Plan. The dilutive effect of these potentially dilutive securities is reflected in diluted earnings per share by application of the treasury stock method. Under the treasury stock method, an increase in the fair market value of our common stock can result in a greater dilutive effect from potentially dilutive securities.
The following table sets forth the computation of basic and diluted net loss per share (in thousands, except per share amounts):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
EPS Numerator:
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(51,464)
|
|
$
|
(90,908)
|
|
$
|
(89,027)
|
|
|
EPS Denominator—Basic and Diluted:
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average common shares outstanding
|
|
|
88,434
|
|
|
87,662
|
|
|
87,288
|
|
|
Net loss per common share:
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted
|
|
$
|
(0.58)
|
|
$
|
(1.04)
|
|
$
|
(1.02)
|
|
During the periods presented, we had securities which could potentially dilute basic loss per share, but were excluded from the computation of diluted net loss per share for all periods presented, as their effect would have been antidilutive. These securities consist of the following (in thousands except per share data):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
Outstanding stock options
|
|
|
|
19,106
|
|
|
16,971
|
|
|
15,532
|
|
|
Warrant to purchase common stock
|
|
|
|
200
|
|
|
200
|
|
|
200
|
|
|
Weighted average exercise price of options
|
|
|
$
|
7.08
|
|
$
|
9.07
|
|
$
|
10.55
|
|
|
Weighted average exercise price of warrant
|
|
|
$
|
6.61
|
|
$
|
6.61
|
|
$
|
6.61
|
|
Recent accounting pronouncements
In August 2014, the FASB issued ASU No. 2014-15—Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern under ASC Subtopic 205-40, Presentation of Financial Statements—Going Concern. ASU No. 2014-15 provides guidance about management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. Management’s evaluation should be based on relevant conditions and events that are known and reasonably knowable at the date that the financial statements are issued (or at the date that the financial statements are available to be issued when applicable). Substantial doubt about an entity’s ability to continue as a going concern exists when relevant conditions and events, considered in the aggregate, indicate that it is probable that the entity will be unable to meet its obligations as they become due within one year after the date that the financial statements are issued (or available to be issued). ASU No. 2014-15 is effective for the annual period ending after December 15, 2016 and early adoption is permitted. We will continue to evaluate the guidance under ASU No. 2014-15 and present the required disclosures within our financial statements at the time of adoption. We plan to adopt this new standard in our annual financial statements for the year ending December 31, 2016 and we believe that the adoption of ASU No. 2014-15 will have no material effect on our financial statement disclosures.
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
In May 2014, the FASB issued ASU No. 2014-09—Revenue from Contracts with Customers, which supersedes the revenue recognition requirements under ASC Topic 605, Revenue Recognition, and most industry-specific guidance under the ASC. The core principle of ASU No. 2014-09 is that an entity should recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 defines a five step process to achieve this core principle and, in doing so, it is possible more judgment and estimates may be required within the revenue recognition process than required under existing U.S. GAAP including identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligation. ASU No. 2014-09 also requires additional disclosures to enable users of financial statements to understand the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts. ASU No. 2014-09 allows for either full retrospective or modified retrospective adoption, and we have not yet determined which approach we will apply. In July 2015, the FASB deferred by one year the effective date of ASU No. 2014-09 with the new effective date beginning after December 15, 2017, and the interim periods within that year and will allow early adoption for all entities as of the original effective date for public business entities, which was annual reporting periods beginning after December 15, 2016. We plan to adopt this new standard on January 1, 2018. We are currently evaluating the potential impact of the adoption of ASU No. 2014-09 on our financial statements and cannot estimate the impact of adoption at this time.
2. SPONSORED RESEARCH AND LICENSE AGREEMENTS
We conduct research and development programs independently and in connection with our corporate collaborators. We are a participant in our collaboration agreement with BMS for the discovery, development and commercialization of cancer immunotherapies based on our small molecule TGF beta receptor kinase inhibitors, as discussed below. Our participation is limited to the Joint Research Committee and the performance of research activities based on billable full-time equivalent fees as specified in the agreement. We do not have ongoing participation obligations under our agreements with Aclaris for the development and commercialization of certain janus kinase (JAK) inhibitors for the treatment of alopecia areata and other dermatological conditions, AstraZeneca (AZ) for the development and commercialization of R256, an inhaled JAK inhibitor, BerGenBio for the development and commercialization of an oncology program, and Daiichi to pursue research related to a specific target from a novel class of drug targets called ligases. Under these agreements, which we entered into in the ordinary course of business, we received or may be entitled to receive upfront cash payments, progress dependent contingent payments on events achieved by such partners and royalties on any net sales of products sold by such partners under the agreements. Total future contingent payments to us under all of these current agreements could exceed $533.6 million if all potential product candidates achieved all of the payment triggering events under all of our current agreements (based on a single product candidate under each agreement). Of this amount, up to $150.5 million relates to the achievement of development events, up to $345.6 million relates to the achievement of regulatory events and up to $37.5 million relates to the achievement of certain commercial or launch events. This estimated future contingent amount does not include any estimated royalties that could be due to us if the partners successfully commercialize any of the licensed products. Future events that may trigger payments to us under the agreements are based solely on our partners’ future efforts and achievements of specified development, regulatory and/or commercial events.
In October 2015, we entered into a non-exclusive license agreement with a third party, pursuant to which we received a payment in the single-digit millions in exchange for granting a non-exclusive license to certain limited intellectual property rights. We concluded that the granting of the license, which was fully delivered to such third party in the fourth quarter of 2015, represents the sole deliverable under this agreement. Accordingly, we have recognized the payment as revenue during the year ended December 31, 2015.
In August 2015, we entered into a license agreement with Aclaris, pursuant to which Aclaris will have exclusive rights and will assume responsibility for the continued development of certain JAK inhibitor compounds for the treatment of alopecia areata and other dermatological conditions. Under the license agreement, we received a
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
noncreditable and non-refundable upfront payment of $8.0 million in September 2015. We are also entitled to receive development and regulatory contingent fees that could exceed $80.0 million for a successful compound approved in certain indications. In addition, we are also eligible to receive tiered royalties on the net sales of any products under the agreement. We concluded that the granting of the license, which has been fully delivered to Aclaris in the third quarter of 2015, represents the sole deliverable under this agreement. Accordingly, we have recognized the $8.0 million payment as revenue during the year ended December 31, 2015.
In February 2015, we entered into a collaboration agreement with BMS for the discovery, development and commercialization of cancer immunotherapies based on our extensive portfolio of small molecule TGF beta receptor kinase inhibitors. Under the collaboration agreement, BMS will have exclusive rights and will be solely responsible for the clinical development and commercialization of any products. Pursuant to the collaboration agreement with BMS, we received a noncreditable and non-refundable upfront payment of $30.0 million in March 2015. We are also entitled to receive development and regulatory contingent fees that could exceed $309.0 million for a successful compound approved in certain indications. In addition, we are also eligible to receive tiered royalties on the net sales of any products from the collaboration. BMS shall also reimburse us for agreed upon costs based on a contractual cost per full-time equivalent employee in connection with the performance of research activities during the research term. Under the collaboration agreement, we were obligated to provide the following deliverables: (i) granting of license rights to our program, (ii) participation in the Joint Research Committee, and (iii) performance of research activities. We concluded that these deliverables are a single unit of accounting as the license does not have stand-alone value apart from the other deliverables. Accordingly, the $30.0 million upfront payment is being recognized ratably as revenue from the effective date of the agreement through September 2016, the end of the estimated research term. We believe that straight-line recognition of this revenue is appropriate as the research is expected to be performed ratably over the research period. During the year ended December 31, 2015, we recognized revenue of $16.6 million and $822,000 relating to the upfront payment and research activities we performed, respectively. As of December 31, 2015, deferred revenue related to the $30.0 million upfront payment was $13.4 million.
3. SIGNIFICANT CONCENTRATIONS
For the year ended December 31, 2015, BMS, Aclaris and another third party accounted for 60%, 28% and 12% of our revenues, respectively. For the year ended December 31, 2014, AZ accounted for all of our revenues. For the year ended December 31, 2013, AZ and Daiichi accounted for 80% and 20% of our revenues, respectively. As of December 31, 2015, we had accounts receivable from BMS of $203,000 relating to the performance of research activities. As of December 31, 2014, we had receivable from AZ of $5.8 million in consideration for AZ’s decision to continue its development of R256 in asthma.
4. STOCK‑BASED COMPENSATION
Total stock‑based compensation expense related to all of our stock‑based awards was as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
Research and development
|
|
|
$
|
4,100
|
|
$
|
4,674
|
|
$
|
3,930
|
|
|
General and administrative
|
|
|
|
3,303
|
|
|
5,113
|
|
|
2,997
|
|
|
Restructuring charges
|
|
|
|
—
|
|
|
—
|
|
|
239
|
|
|
Total stock-based compensation expense
|
|
|
$
|
7,403
|
|
$
|
9,787
|
|
$
|
7,166
|
|
In December 2014, we entered into a severance agreement with our former CEO. As part of the severance arrangement we offered, we extended the date to which our former CEO had the right to exercise his vested options within 90 days from his termination date as was stipulated under his option agreement to the end of the contractual term
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
of the options, of which the remaining contractual term for the most recently granted options is nine years. In addition, we also accelerated the vesting period of certain of his unvested stock options. As a result of these modifications, we recorded incremental stock-based compensation expense of approximately $1.5 million in the fourth quarter of 2014 (see Note 11). This amount is included as part of “General and administrative expense” in the accompanying Statement of Operations.
In September 2013, we announced that we had reduced our workforce by 18%, or 30 positions, in connection with efforts to prioritize projects and conserve our working capital. As part of the severance arrangement we offered the terminated employees, we extended the date to which the terminated employees had to exercise their vested options to June 30, 2014, rather than 90 days from the termination date as was stipulated under the employee’s option agreements. In addition, we also accelerated the vesting period of certain unvested stock options for one terminated employee. As a result of these modifications, we recorded non‑cash stock‑based compensation expense of $239,000 in the third quarter of 2013. This expense was classified under “Restructuring expense” in the accompanying Statements of Operations.
Employee Stock Option Plans
We have three stock option plans, our 2011 Equity Incentive Plan (2011 Plan), 2000 Equity Incentive Plan (2000 Plan) and 2000 Non‑Employee Directors Stock Option Plan (Directors’ Plan), that provide for granting to our officers, directors and all other employees and consultants options to purchase shares of our common stock. Options granted under our 2011 Plan expire no later than ten years from the date of grant. Options may be granted with different vesting terms from time to time, ranging from zero to five years. As of December 31, 2015, a total of 10,864,592 shares of common stock were authorized for issuance under the 2011 Plan. There were 214,295 options to purchase shares exercised during the year ended December 31, 2015 under the 2011 Plan. Options under the 2000 Plan may be granted with different vesting terms from time to time, ranging from zero to five years. As of December 31, 2015, a total of 12,299,675 shares of common stock were authorized for issuance under the 2000 Plan. There were no options to purchase shares exercised during the year ended December 31, 2015 under the 2000 Plan. Options under the Directors’ Plan may be granted for a maximum term 10 years. The exercise price of options under the Directors’ Plan is equal to the fair market value of the common stock on the date of grant. As of December 31, 2015, a total of 1,188,182 shares of common stock were authorized for issuance under the Directors’ Plan. There were no options to purchase shares exercised during the year ended December 31, 2015 under the Directors’ Plan.
Pursuant to FASB ASC 718, we are required to estimate the amount of expected forfeitures when calculating compensation costs. We estimated the forfeiture rate using our historical experience of actual forfeitures. We adjust our stock‑based compensation expense as actual forfeitures occur, review our estimated forfeiture rates each quarter and make changes to our estimate as appropriate.
The fair value of each option award is estimated on the date of grant using the Black‑Scholes option pricing model. We have segregated option awards into the following three homogenous groups for the purposes of determining fair values of options: officers and directors, all other employees, and consultants.
We determined weighted‑average valuation assumptions separately for each of these groups as follows:
|
·
| |
Volatility—We estimated volatility using the historical share price performance over the expected life of the option up to the point where we have historical market data. We also considered other factors, such as implied volatility, our current clinical trials and other company activities that may affect the volatility of our stock in the future. We determined that at this time historical volatility is more indicative of our expected future stock performance than implied volatility. |
|
·
| |
Expected term—For options granted to consultants, we use the contractual term of the option, which is generally ten years, for the initial valuation of the option and the remaining contractual term of the option |
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
for the succeeding periods. We worked with various historical data to determine the applicable expected term for each of the other option groups. This data included: (1) for exercised options, the term of the options from option grant date to exercise date; (2) for cancelled options, the term of the options from option grant date to cancellation date, excluding nonvested option forfeitures; and (3) for options that remained outstanding at the balance sheet date, the term of the options from option grant date to the end of the reporting period and the estimated remaining term of the options. The consideration and calculation of the above data gave us reasonable estimates of the expected term for each employee group. We also considered the vesting schedules of the options granted and factors surrounding exercise behavior of the option groups, our current market price and company activity that may affect our market price. In addition, we considered the optionee type (i.e., officers and directors or all other employees) and other factors that may affect the expected term of the option. |
|
·
| |
Risk‑free interest rate—The risk‑free interest rate is based on U.S. Treasury constant maturity rates with similar terms to the expected term of the options for each option group. |
|
·
| |
Dividend yield—The expected dividend yield is 0% as we have not paid and do not expect to pay dividends in the future. |
The following table summarizes the weighted‑average assumptions relating to options granted pursuant to our equity incentive plans for the years ended December 31, 2015, 2014 and 2013:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended
|
|
|
|
|
|
December 31,
|
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
Risk-free interest rate
|
|
|
1.8
|
%
|
2.2
|
%
|
1.1
|
%
|
|
Expected term (in years)
|
|
|
6.5
|
|
6.5
|
|
5.4
|
|
|
Dividend yield
|
|
|
0.0
|
%
|
0.0
|
%
|
0.0
|
%
|
|
Expected volatility
|
|
|
65.0
|
%
|
74.4
|
%
|
72.2
|
%
|
The exercise price of stock options is determined to be the market price of our common stock on the date immediately preceding the date of grant. These stock options become exercisable at varying dates and generally expire ten years from the date of grant. At December 31, 2015, options to purchase 5,245,977 shares of common stock were available for grant and 24,352,449 reserved shares of common stock were available for future issuance under our stock option plans.
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
Stock‑Based Compensation Award Activity
Option activity under our equity incentive plans was as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining
|
|
|
|
|
|
|
|
Shares Available
|
|
Number of Shares
|
|
Weighted-Average
|
|
Contractual Term
|
|
Aggregate
|
|
|
|
|
For Grant
|
|
Underlying Options
|
|
Exercise Price
|
|
(in years)
|
|
Intrinsic Value
|
|
|
Outstanding at January 1, 2013
|
|
3,198,586
|
|
13,604,377
|
|
$
|
11.52
|
|
|
|
|
|
|
|
Authorized for grant
|
|
7,775,000
|
|
—
|
|
|
|
|
|
|
|
|
|
|
Granted
|
|
(3,140,956)
|
|
3,140,956
|
|
$
|
5.46
|
|
|
|
|
|
|
|
Exercised
|
|
—
|
|
—
|
|
$
|
—
|
|
|
|
|
|
|
|
Cancelled
|
|
1,213,131
|
|
(1,213,131)
|
|
$
|
8.26
|
|
|
|
|
|
|
|
Outstanding at December 31, 2013
|
|
9,045,761
|
|
15,532,202
|
|
$
|
10.55
|
|
|
|
|
|
|
|
Authorized for grant
|
|
—
|
|
—
|
|
|
|
|
|
|
|
|
|
|
Granted
|
|
(3,467,120)
|
|
3,467,120
|
|
$
|
3.39
|
|
|
|
|
|
|
|
Exercised
|
|
—
|
|
(11,219)
|
|
$
|
3.44
|
|
|
|
|
|
|
|
Cancelled
|
|
2,017,430
|
|
(2,017,430)
|
|
$
|
10.75
|
|
|
|
|
|
|
|
Outstanding at December 31, 2014
|
|
7,596,071
|
|
16,970,673
|
|
$
|
9.07
|
|
|
|
|
|
|
|
Authorized for grant
|
|
—
|
|
—
|
|
|
|
|
|
|
|
|
|
|
Granted
|
|
(3,875,170)
|
|
3,875,170
|
|
$
|
2.28
|
|
|
|
|
|
|
|
Exercised
|
|
—
|
|
(214,295)
|
|
$
|
2.75
|
|
|
|
|
|
|
|
Cancelled
|
|
1,525,076
|
|
(1,525,076)
|
|
$
|
17.56
|
|
|
|
|
|
|
|
Outstanding at December 31, 2015
|
|
5,245,977
|
|
19,106,472
|
|
$
|
7.08
|
|
5.85
|
|
$
|
3,053,499
|
|
|
Vested and expected to vest at December 31, 2015
|
|
|
|
18,958,691
|
|
$
|
7.12
|
|
|
|
|
|
|
|
Exercisable at December 31, 2015
|
|
|
|
15,089,131
|
|
$
|
8.24
|
|
5.08
|
|
$
|
865,450
|
|
|
Exercisable at December 31, 2014
|
|
|
|
14,243,893
|
|
$
|
10.09
|
|
|
|
|
|
|
|
Exercisable at December 31, 2013
|
|
|
|
13,246,612
|
|
$
|
11.44
|
|
|
|
|
|
|
Of the 3,875,170 common stock options granted during 2015, 1,175,000 shares were related to performance-based stock option awards which will vest upon the achievement of a corporate performance-based milestone related to the progress of the Phase 3 clinical program of fostamatinib in ITP. Of the 3,467,120 common stock options granted during 2014, 950,000 shares were related to performance-based stock option awards, of which only 700,000 shares remain outstanding due to the cancellation of the 250,000 shares in the fourth quarter of 2014. These remaining shares will vest upon the achievement of certain corporate performance-based milestones related to the progress and success of the Phase 3 clinical program of fostamatinib in ITP. Weighted‑average grant date fair value of options granted during 2015, 2014 and 2013 was $1.40, $2.32 and $3.34, respectively.
The aggregate intrinsic value of the stock options in the table above is calculated as the difference between the exercise price of the underlying awards and the quoted price of our common stock for the options that were in‑the‑money at December 31, 2015. At December 31, 2015 and 2014, we had 4,017,340 and 2,726,779, respectively, of nonvested stock options, with approximately $2.2 million and $24,000 intrinsic value at December 31, 2015 and 2014, respectively. During the years ended December 31, 2015 and 2014, aggregate intrinsic value of options exercised under our stock option plans was approximately $252,000 and $10,000, respectively, determined as of the date of the stock option exercise.
As of December 31, 2015, there was approximately $4.3 million of total unrecognized compensation cost, net of estimated forfeitures, related to nonvested stock‑based compensation arrangements granted under our stock option plans and approximately $276,000 of total unamortized compensation cost related to our Purchase Plan. The unamortized compensation cost related to our stock option plans and our Purchase Plan is expected to be recognized over a weighted‑ average period of approximately 0.9 years and 0.4 years, respectively. For the years ended December 31, 2015
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
and 2014, there were 2,326,021 and 2,456,622 shares vested, respectively, with weighted‑average exercise price of $3.13 and $4.74, respectively.
Details of our stock options by exercise price are as follows as of December 31, 2015:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options Outstanding
|
|
Options Exercisable
|
|
|
|
|
Number of
|
|
Weighted-Average
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding
|
|
Remaining
|
|
Weighted-Average
|
|
Number of
|
|
Weighted-Average
|
|
|
Exercise Price
|
|
Options
|
|
Contractual Life (in years)
|
|
Exercise Price
|
|
Options
|
|
Exercise Price
|
|
|
$1.68 - $2.14
|
|
3,398,170
|
|
8.99
|
|
$
|
2.14
|
|
965,799
|
|
$
|
2.14
|
|
|
$2.72 - $3.59
|
|
3,818,711
|
|
7.99
|
|
|
3.43
|
|
2,419,487
|
|
|
3.40
|
|
|
$3.64 - $6.51
|
|
3,613,435
|
|
5.53
|
|
|
6.31
|
|
3,432,883
|
|
|
6.37
|
|
|
$6.55 - $7.40
|
|
2,490,644
|
|
4.24
|
|
|
6.81
|
|
2,490,644
|
|
|
6.81
|
|
|
$7.53 - $9.62
|
|
3,392,461
|
|
4.83
|
|
|
8.70
|
|
3,387,320
|
|
|
8.70
|
|
|
$9.74 - $26.45
|
|
2,393,051
|
|
1.58
|
|
|
19.11
|
|
2,392,999
|
|
|
19.11
|
|
|
$1.68 - $26.45
|
|
19,106,472
|
|
5.85
|
|
|
7.08
|
|
15,089,132
|
|
|
8.24
|
|
Employee Stock Purchase Plan
Our Employee Stock Purchase Plan (Purchase Plan) permits eligible employees to purchase common stock at a discount through payroll deductions during defined offering periods. The price at which the stock is purchased is equal to the lesser of 85% of the fair market value of the common stock on the first day of the offering or 85% of the fair market value of our common stock on the purchase date. The initial offering period commenced on the effective date of our initial public offering. We issued 576,537, 505,877 and 383,717 shares of common stock during 2015, 2014 and 2013, respectively, pursuant to the Purchase Plan at an average price of $2.03, $2.24 and $2.74, respectively. For 2015, 2014 and 2013, the weighted average fair value of awards granted under our Purchase Plan was $1.05, $1.42 and $2.05, respectively. As of December 31, 2015, we had 3,001,616 reserved shares of common stock available for future issuance under the Purchase Plan.
The fair value of awards granted under our Purchase Plan is estimated on the date of grant using the Black‑Scholes option pricing model, which uses weighted‑ average assumptions. Our Purchase Plan provides for a twenty‑four month offering period comprised of four six‑month purchase periods with a look‑back option. A look‑back option is a provision in our Purchase Plan under which eligible employees can purchase shares of our common stock at a price per share equal to the lesser of 85% of the fair market value on the first day of the offering period or 85% of the fair market value on the purchase date. Our Purchase Plan also includes a feature that provides for a new offering period to begin when the fair market value of our common stock on any purchase date during an offering period falls below the fair market value of our common stock on the first day of such offering period. This feature is called a “reset.” Participants are automatically enrolled in the new offering period. We had a “reset” on January 2, 2014 because the fair market value of our stock on December 31, 2013 was lower than the fair market value of our stock on July 1, 2013, the first day of the offering period. We applied modification accounting in accordance with ASC Topic No. 718, Stock Compensation, to determine the incremental fair value associated with this Purchase Plan “reset” and will recognize the related stock‑based compensation expense according to FASB ASC Subtopic No. 718‑50, Employee Share Purchase Plan. The total incremental fair value for this Purchase Plan “reset” was approximately $577,000, that will be recognized from January 2, 2014 to December 31, 2015. On January 2, 2015, we had another “reset” because the fair market value of our stock on December 31, 2014 was lower than the fair market value of our stock on July 1, 2014, the first day of another offering period. We applied modification accounting in accordance with the relevant guidance and determined that the incremental fair value associated with this Purchase Plan “reset” was approximately $792,000 that will be recognized from January 2, 2015 to December 31, 2016.
The following table summarizes the weighted‑average assumptions related to our Purchase Plan for the years ended December 31, 2015, 2014 and 2013. Expected volatilities for our Purchase Plan are based on the two‑year
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
historical volatility of our stock. Expected term represents the weighted‑ average of the purchase periods within the offering period. The risk‑free interest rate for periods within the expected term is based on U.S. Treasury constant maturity rates.
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended
|
|
|
|
|
December 31,
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
Risk-free interest rate
|
|
0.6
|
%
|
0.3
|
%
|
0.2
|
%
|
|
Expected term (in years)
|
|
1.5
|
|
1.7
|
|
1.4
|
|
|
Dividend yield
|
|
0.0
|
%
|
0.0
|
%
|
0.0
|
%
|
|
Expected volatility
|
|
61.2
|
%
|
66.0
|
%
|
64.4
|
%
|
5. CASH, CASH EQUIVALENTS AND SHORT-TERM INVESTMENTS
Cash, cash equivalents and short-term investments consist of the following (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2015
|
|
2014
|
|
|
Checking account
|
|
$
|
2,118
|
|
$
|
175
|
|
|
Money market funds
|
|
|
26,291
|
|
|
10,027
|
|
|
U. S. treasury bills
|
|
|
9,048
|
|
|
2,010
|
|
|
Government-sponsored enterprise securities
|
|
|
48,613
|
|
|
45,786
|
|
|
Corporate bonds and commercial paper
|
|
|
40,206
|
|
|
85,161
|
|
|
|
|
$
|
126,276
|
|
$
|
143,159
|
|
|
Reported as:
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
43,456
|
|
$
|
15,203
|
|
|
Short-term investments
|
|
|
82,820
|
|
|
127,956
|
|
|
|
|
$
|
126,276
|
|
$
|
143,159
|
|
Cash equivalents and short-term investments included the following securities with gross unrealized gains and losses (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross
|
|
Gross
|
|
|
|
|
|
|
|
Amortized
|
|
Unrealized
|
|
Unrealized
|
|
|
|
|
|
December 31, 2015
|
|
Cost
|
|
Gains
|
|
Losses
|
|
Fair Value
|
|
|
U. S. treasury bills
|
|
$
|
9,061
|
|
$
|
—
|
|
$
|
(13)
|
|
$
|
9,048
|
|
|
Government-sponsored enterprise securities
|
|
|
48,643
|
|
|
1
|
|
|
(31)
|
|
|
48,613
|
|
|
Corporate bonds and commercial paper
|
|
|
40,207
|
|
|
11
|
|
|
(12)
|
|
|
40,206
|
|
|
Total
|
|
$
|
97,911
|
|
$
|
12
|
|
$
|
(56)
|
|
$
|
97,867
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross
|
|
Gross
|
|
|
|
|
|
|
|
Amortized
|
|
Unrealized
|
|
Unrealized
|
|
|
|
|
|
December 31, 2014
|
|
Cost
|
|
Gains
|
|
Losses
|
|
Fair Value
|
|
|
U. S. treasury bills
|
|
$
|
2,010
|
|
$
|
—
|
|
$
|
—
|
|
$
|
2,010
|
|
|
Government-sponsored enterprise securities
|
|
|
45,793
|
|
|
4
|
|
|
(11)
|
|
|
45,786
|
|
|
Corporate bonds and commercial paper
|
|
|
85,161
|
|
|
21
|
|
|
(21)
|
|
|
85,161
|
|
|
Total
|
|
$
|
132,964
|
|
$
|
25
|
|
$
|
(32)
|
|
$
|
132,957
|
|
As of December 31, 2015, the contractual maturities of our cash equivalents and short-term investments were (in thousands):
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
|
|
|
|
|
|
|
|
|
|
|
|
Years to Maturity
|
|
|
|
|
|
|
|
After One Year
|
|
|
|
|
Within
|
|
Through
|
|
|
|
|
One Year
|
|
Two Years
|
|
|
U. S. treasury bills
|
|
$
|
4,753
|
|
$
|
4,295
|
|
|
Government-sponsored enterprise securities
|
|
|
48,613
|
|
|
—
|
|
|
Corporate bonds and commercial paper
|
|
|
40,206
|
|
|
—
|
|
|
|
|
$
|
93,572
|
|
$
|
4,295
|
|
As of December 31, 2015, our cash equivalents and short-term investments had a weighted‑average time to maturity of approximately 118 days. We view our short-term investments portfolio as available for use in current operations. Accordingly, we have classified certain securities as short-term investments on our balance sheet even though the stated maturity date of these securities may be more than one year from the current balance sheet date. We have the ability to hold all investments as of December 31, 2015 through their respective maturity dates. At December 31, 2015, we had no investments that had been in a continuous unrealized loss position for more than 12 months. As of December 31, 2015, a total of 43 individual securities had been in an unrealized loss position for 12 months or less and the losses were deemed to be temporary. The gross unrealized losses above were caused by interest rate increases. No significant facts or circumstances have arisen to indicate that there has been any deterioration in the creditworthiness of the issuers of the securities held by us. Based on our review of these securities, including the assessment of the duration and severity of the unrealized losses and our ability and intent to hold the investments until maturity, there were no other-than-temporary impairments for these securities at December 31, 2015.
The following table shows the fair value and gross unrealized losses of our investments in individual securities that are in an unrealized loss position, aggregated by investment category (in thousands):
|
|
|
|
|
|
|
|
|
|
December 31, 2015
|
|
Fair Value
|
|
Unrealized Losses
|
|
|
U. S. treasury bills
|
|
$
|
8,294
|
|
$
|
(13)
|
|
|
Government-sponsored enterprise securities
|
|
|
41,266
|
|
|
(31)
|
|
|
Corporate bonds and commercial paper
|
|
|
17,826
|
|
|
(12)
|
|
|
Total
|
|
$
|
67,386
|
|
$
|
(56)
|
|
6. FAIR VALUE
Under FASB ASC 820, Fair Value Measurements and Disclosures, fair value is defined as the price at which an asset could be exchanged or a liability transferred in a transaction between knowledgeable, willing parties in the principal or most advantageous market for the asset or liability. Where available, fair value is based on observable market prices or parameters or derived from such prices or parameters. Where observable prices or parameters are not available, valuation models are applied.
Assets and liabilities recorded at fair value in our financial statements are categorized based upon the level of judgment associated with the inputs used to measure their fair value. Hierarchical levels directly related to the amount of subjectivity associated with the inputs to fair valuation of these assets and liabilities, are as follows:
Level 1—Inputs are unadjusted, quoted prices in active markets for identical assets at the reporting date. Active markets are those in which transactions for the asset or liability occur in sufficient frequency and volume to provide pricing information on an ongoing basis.
The fair valued assets we hold that are generally included under this Level 1 are money market securities where fair value is based on publicly quoted prices.
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
Level 2—Are inputs, other than quoted prices included in Level 1, that are either directly or indirectly observable for the asset or liability through correlation with market data at the reporting date and for the duration of the instrument’s anticipated life.
The fair valued assets we hold that are generally assessed under Level 2 included government‑sponsored enterprise securities, U. S. treasury bills and corporate bonds and commercial paper. We utilize third party pricing services in developing fair value measurements where fair value is based on valuation methodologies such as models using observable market inputs, including benchmark yields, reported trades, broker/dealer quotes, bids, offers and other reference data. We use quotes from external pricing service providers and other on‑line quotation systems to verify the fair value of investments provided by our third party pricing service providers. We review independent auditor’s reports from our third party pricing service providers particularly regarding the controls over pricing and valuation of financial instruments and ensure that our internal controls address certain control deficiencies, if any, and complementary user entity controls are in place.
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities and which reflect management’s best estimate of what market participants would use in pricing the asset or liability at the reporting date. Consideration is given to the risk inherent in the valuation technique and the risk inherent in the inputs to the model.
We do not have fair valued assets classified under Level 3.
Fair Value on a Recurring Basis
Financial assets measured at fair value on a recurring basis are categorized in the tables below based upon the lowest level of significant input to the valuations (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Assets at Fair Value as of December 31, 2015
|
|
|
|
|
Level 1
|
|
Level 2
|
|
Level 3
|
|
Total
|
|
|
Money market funds
|
|
$
|
26,291
|
|
$
|
—
|
|
$
|
—
|
|
$
|
26,291
|
|
|
U. S. treasury bills
|
|
|
—
|
|
|
9,048
|
|
|
—
|
|
|
9,048
|
|
|
Government-sponsored enterprise securities
|
|
|
—
|
|
|
48,613
|
|
|
—
|
|
|
48,613
|
|
|
Corporate bonds and commercial paper
|
|
|
—
|
|
|
40,206
|
|
|
—
|
|
|
40,206
|
|
|
Total
|
|
$
|
26,291
|
|
$
|
97,867
|
|
$
|
—
|
|
$
|
124,158
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Assets at Fair Value as of December 31, 2014
|
|
|
|
|
Level 1
|
|
Level 2
|
|
Level 3
|
|
Total
|
|
|
Money market funds
|
|
$
|
10,027
|
|
$
|
—
|
|
$
|
—
|
|
$
|
10,027
|
|
|
U. S. treasury bills
|
|
|
—
|
|
|
2,010
|
|
|
—
|
|
|
2,010
|
|
|
Government-sponsored enterprise securities
|
|
|
—
|
|
|
45,786
|
|
|
—
|
|
|
45,786
|
|
|
Corporate bonds and commercial paper
|
|
|
—
|
|
|
85,161
|
|
|
—
|
|
|
85,161
|
|
|
Total
|
|
$
|
10,027
|
|
$
|
132,957
|
|
$
|
—
|
|
$
|
142,984
|
|
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
7. PROPERTY AND EQUIPMENT
Property and equipment consists of the following (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2015
|
|
2014
|
|
|
Laboratory equipment
|
|
$
|
17,892
|
|
$
|
23,228
|
|
|
Computer and software
|
|
|
1,189
|
|
|
1,225
|
|
|
Furniture and equipment
|
|
|
682
|
|
|
1,177
|
|
|
Total property and equipment
|
|
$
|
19,763
|
|
$
|
25,630
|
|
|
Less accumulated depreciation and amortization
|
|
|
(18,150)
|
|
|
(23,121)
|
|
|
Property and equipment, net
|
|
$
|
1,613
|
|
$
|
2,509
|
|
During 2015 and 2014, we disposed of approximately $6.5 million and $1.6 million, respectively, of fully depreciated assets.
Total depreciation and amortization expense was $1.4 million, $2.4 million and $2.6 million for the years ended December 31, 2015, 2014 and 2013, respectively.
8. LONG‑TERM OBLIGATIONS
We currently lease our research and office space under a noncancelable lease agreement with our landlord, HCP BTC, LLC (formerly known as Slough BTC, LLC) which expires in 2018. The lease term provides for renewal option for up to two additional periods of five years each, and rental payments on a graduated scale. We determined our existing lease agreement to be an operating lease and recognize rent expense on a straight‑line basis over the lease period.
In December 2014, we entered into a sublease agreement with an unrelated third party to occupy a portion of our research and office space. We expect to receive over $5.0 million in future sublease income (excluding our subtenant’s share of facilities operating expenses) over the remaining term of the sublease. In connection with this sublease, we recognized a loss on sublease of $9.3 million during the fourth quarter of 2014. We record rent expense on a straight-line basis for our lease, net of sublease income, wherein such arrangements contain scheduled rent increases over the term of the lease and sublease, respectively. For our sublease arrangement which we classified as an operating lease, our loss on the sublease was comprised of the present value of our future payments to our landlord less the present value of our future rent payments expected from our subtenant over the term of the sublease. Further, in conjunction with our facilities lease, we have previously issued to our landlord warrants to purchase our common stock. We have previously capitalized the fair value of these warrants at issuance as part of our other long-term assets and they are being amortized over the term of our lease. As a result of the sublease agreement that we entered into in December 2014, we included approximately $265,000 representing the unamortized portion of the warrant fair value attributable to the sublet space in the determination of our loss on sublease (see Note 9). The liability arising from this sublease agreement was determined using a credit-adjusted risk-free rate to discount the estimated future net cash flows.
The changes in the liability related to the sublease agreement during the year ended December 31, 2015 were as follows (in thousands):
|
|
|
|
|
|
|
Balance at January 1, 2015
|
|
$
|
9,269
|
|
|
Accretion of deferred liability
|
|
|
559
|
|
|
Amortization of deferred liability
|
|
|
(3,363)
|
|
|
Balance at December 31, 2015
|
|
$
|
6,465
|
|
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
At December 31, 2015, future minimum lease payments and obligations under our noncancelable operating lease, net of sublease receipts, were as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating
|
|
Sublease
|
|
|
|
|
|
For years ending December 31,
|
|
Lease
|
|
Receipts
|
|
Net
|
|
|
2016
|
|
$
|
15,530
|
|
$
|
(2,771)
|
|
$
|
12,759
|
|
|
2017
|
|
|
16,153
|
|
|
(2,854)
|
|
|
13,299
|
|
|
2018
|
|
|
1,351
|
|
|
(196)
|
|
|
1,155
|
|
|
Total minimum payments required
|
|
$
|
33,034
|
|
$
|
(5,821)
|
|
$
|
27,213
|
|
Rent expense under our operating lease amounted to approximately $8.9 million (net of sublease income, subtenant’s share of certain facilities operating expense and amortization of deferred liability in the aggregate total of $6.3 million), $15.1 million and $14.8 million for the years ended December 31, 2015, 2014 and 2013, respectively.
9. STOCKHOLDERS’ EQUITY
Preferred Stock
We are authorized to issue 10,000,000 shares of preferred stock. As of December 31, 2015 and 2014, there were no issued and outstanding shares of preferred stock. Our board of directors is authorized to fix or alter the designation, powers, preferences and rights of the shares of each series of preferred shares, and the qualifications, limitations or restrictions of any wholly unissued shares, to establish from time to time the number of shares constituting any such series, and to increase or decrease the number of shares, if any.
Warrants
In conjunction with the facilities lease entered into in May 2001, we issued a warrant to the lessor to purchase 16,666 shares of our common stock at an exercise price of $80.21 per share, a 15% premium to market at the time of issuance. This warrant expired unexercised in May 2006. The fair market value of this warrant, as determined using the Black‑Scholes valuation model, was approximately $683,000. This amount has been capitalized in other long‑term assets and is being amortized into expense over the life of the lease. As of December 31, 2015, approximately $58,000 remained to be amortized over the remaining term of the lease.
In conjunction with the facilities lease amendment in October 2002, we issued a warrant to the lessor to purchase 55,555 shares of our common stock at an exercise price of $17.73 per share. The warrant expired unexercised in October 2007. The fair value of this warrant, as determined using the Black‑Scholes valuation model, was approximately $565,000. This amount has been capitalized in other long‑term assets and is being amortized into expense over the life of the lease. As of December 31, 2015, approximately $48,000 remained to be amortized over the term of the lease.
In conjunction with the facilities lease amendment in July 2006, we issued a warrant to the lessor to purchase 100,000 shares of our common stock at an exercise price of $10.57 per share. The fair value of this warrant, as determined using the Black‑Scholes valuation model, was approximately $801,000. This amount has been included in other long‑term assets and is being amortized into expense over the term of the lease. As of December 31, 2015, approximately $88,000 remained to be amortized over the term of the lease. The lease agreement was further amended in March 2009. The lease amendment provided for the cancellation of the abovementioned warrant to purchase 100,000 shares of common stock and the issuance of a new warrant granting our landlord the right to purchase 200,000 shares of common stock. The exercise price per share of the new warrant is $6.61. The new warrant is outstanding as of December 31, 2015 and remains exercisable at any time up to February 2016. We applied modification accounting in
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
2009 and determined the fair value of this warrant using the Black‑Scholes valuation model. The incremental fair value of the new warrant as a result of the modification is $616,000. This amount has been included in other long‑term assets and is being amortized into expense over the term of the lease. As of December 31, 2015, approximately $89,000 remained to be amortized over the term of the lease.
As discussed in Note 8, as a result of the sublease agreement that we entered into in December 2014, we included approximately $265,000 representing the unamortized portion of the above fair value of warrants attributable to the sublet space in the determination of our loss on sublease during the year ended December 31, 2014.
Controlled Equity Offering
In August 2015, we entered into a Controlled Equity OfferingSM Sales Agreement with Cantor, as sales agent, pursuant to which we may sell, through Cantor, up to an aggregate of $30.0 million in shares of our common stock. All sales of our common stock will be made pursuant to a shelf registration statement that was declared effective by the Securities and Exchange Commission (SEC) on July 13, 2015. Cantor is acting as our sole sales agent for any sales made under the Sales Agreement for a low single-digit commission on gross proceeds. The common stock is being sold at prevailing market prices at the time of the sale. Unless otherwise terminated earlier, the Controlled Equity OfferingSM Sales Agreement continues until all shares available under the agreement have been sold. During the year ended December 31, 2015, approximately, 1,722,312 shares of our common stock were sold under the Sales Agreement with aggregate net proceeds of $5.5 million. At December 31, 2015, we had approximately $24.3 million in shares of our common stock registered for sale under the Sales Agreement.
10. INCOME TAXES
For the years ended December 31, 2015, 2014 and 2013, our loss before income taxes was from domestic operations. For the years ended December 31, 2015, 2014 and 2013, we did not record a provision for income taxes due to our net loss.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets are as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2015
|
|
2014
|
|
|
Deferred tax assets
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards
|
|
$
|
276,740
|
|
$
|
268,400
|
|
|
Orphan drug and research and development credits
|
|
|
36,387
|
|
|
31,735
|
|
|
Deferred compensation
|
|
|
22,647
|
|
|
28,408
|
|
|
Capitalized research and development expenses
|
|
|
1,761
|
|
|
20,312
|
|
|
Other, net
|
|
|
4,966
|
|
|
7,876
|
|
|
Total deferred tax assets
|
|
|
342,501
|
|
|
356,731
|
|
|
Valuation allowance
|
|
|
(342,501)
|
|
|
(356,731)
|
|
|
Net deferred tax assets
|
|
$
|
—
|
|
$
|
—
|
|
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
The reconciliation of the statutory federal income tax rate to the effective tax rate was as follows:
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2015
|
|
2014
|
|
2013
|
|
|
Federal statutory tax rate
|
|
(34.0)
|
%
|
(34.0)
|
%
|
(34.0)
|
%
|
|
Valuation allowance
|
|
31.3
|
%
|
32.3
|
%
|
37.9
|
%
|
|
Other, net
|
|
2.7
|
%
|
1.7
|
%
|
(3.9)
|
%
|
|
Effective tax rate
|
|
0.0
|
%
|
0.0
|
%
|
0.0
|
%
|
In general, under Section 382 of the Internal Revenue Code (Section 382), a corporation that undergoes an ownership change is subject to limitations on its ability to utilize its pre‑change net operating loss carryovers and tax credits to offset future taxable income. Our existing net operating loss carryforwards and tax credits are subject to limitations arising from ownership changes which occurred in previous periods. We finalized our analysis of potential ownership changes and concluded our Section 382 owner shift analysis during the year ended December 31, 2012. We have updated our net operating loss carryforwards to reflect the results of the Section 382 owner shift analysis as of December 31, 2015. We did not experience any significant changes in ownership in 2015 and 2014. Future changes in our stock ownership, some of which are outside of our control, could result in an ownership change under Section 382 and result in additional limitations.
As of December 31, 2015, we had net operating loss carryforwards for federal income tax purposes of approximately $764.7 million, which expire beginning in the year 2019 and state net operating loss carryforwards of approximately $424.8 million, which expire beginning in the year 2016. We had elected the three-factor apportionment formula pursuant to the Multistate Tax Compact, or MTC in determining the state net operating loss carryforwards for 2013 and 2014. In December 2015, the California Supreme Court overturned the California Appellate court decision on The Gillette Company et al. v. California Franchise Tax Board. The court held that the taxpayers couldn’t elect an evenly weighted, three-factor apportionment formula pursuant to the MTC. As a result of the California Supreme Court decision, we reduced our deferred tax assets and offsetting valuation allowance related to the California NOL calculated in 2013 and 2014 pursuant to the MTC election.
We have general business credits of approximately $25.6 million, which will expire beginning in 2023, if not utilized, and is comprised of research and development credits and orphan drug credits. We also have state research and development tax credits of approximately$23.6 million, which have no expiration date.
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance decreased by approximately $14.2 million and increased by approximately $62.6 million for the years ended December 31, 2015 and 2014, respectively.
Included in the valuation allowance balance at December 31, 2015 and 2014 is approximately $2.5 million of tax deductions related to the exercise of stock options prior to the adoption of ASC 718 which have not reflected as an expense for financial reporting purposes. Accordingly, any future reduction in the valuation allowance relating to this amount will be credited directly to equity and not reflected as an income tax benefit in the statement of operations. As a result of certain realization requirements, the table of deferred tax assets and liabilities shown above does not include loss carryforward tax assets of approximately $1.7 million at December 31, 2015 and 2014 that arose directly from (or the use of which was postponed by) tax deductions related to stock‑based compensation expense in excess of compensation expense recognized for financial reporting. Equity will be increased by approximately $1.7 million if and when such deferred tax assets are ultimately realized.
Table of Contents
Rigel Pharmaceuticals, Inc.
NOTES TO FINANCIAL STATEMENTS (Continued)
The following table summarizes the activity related to our gross unrecognized tax benefits (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2015
|
|
2014
|
|
|
Balance at the beginning of the year
|
|
$
|
5,374
|
|
$
|
5,001
|
|
|
Increase related to prior year tax positions
|
|
|
11,332
|
|
|
—
|
|
|
Increase related to current year tax positions
|
|
|
572
|
|
|
373
|
|
|
Balance at the end of the year
|
|
$
|
17,278
|
|
$
|
5,374
|
|
Included in the balance of unrecognized tax benefits at December 31, 2015 and 2014, respectively, are $12.2 million and $4.3 million of tax benefits that, if recognized, would result in adjustments to other tax accounts, primarily deferred taxes. No income tax benefit would be realized due to the Company’s valuation allowance position. We do not anticipate a significant change to the unrecognized tax benefits over the next twelve months.
We are subject to taxation in the United States and in California. Because of net operating loss and research credit carryovers, substantially all of our tax years remain open to examination.
Our policy is that we recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. We currently have no tax positions that would be subject to interest or penalties.
11. SEVERANCE AGREEMENT WITH FORMER CEO
In 2014, we entered into a severance agreement with our former CEO pursuant to his resignation as CEO and member of the Board of Directors effective November 20, 2014, and his retirement effective December 31, 2014. The severance agreement provides for cash severance payments of $1.1 million payable in installments over a duration of 18 months beginning on January 1, 2015, which is included as part of the Accrued Compensation account in the Balance Sheets. Also as part of the severance arrangement, we extended the date to which our former CEO had the right to exercise his vested options. In addition, we also accelerated the vesting of certain of his unvested stock options (refer to Note 4). The change in the severance liability to our former CEO during the year ended December 31, 2015 was as follows (in thousands):
|
|
|
|
|
|
|
Balance at January 1, 2015
|
|
$
|
1,091
|
|
|
Payments during the year
|
|
|
(724)
|
|
|
Balance at December 31, 2015
|
|
$
|
367
|
|
12. SELECTED QUARTERLY FINANCIAL DATA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2015
|
|
Year Ended December 31, 2014
|
|
|
|
|
Q1
|
|
Q2
|
|
Q3
|
|
Q4
|
|
Q1
|
|
Q2
|
|
Q3
|
|
Q4
|
|
|
|
|
(unaudited, in thousands, except per share amounts)
|
|
|
Revenue
|
|
$
|
2,178
|
|
$
|
5,184
|
|
$
|
12,996
|
|
$
|
8,537
|
|
$
|
—
|
|
$
|
—
|
|
$
|
—
|
|
$
|
8,250
|
|
|
Net loss
|
|
$
|
(18,193)
|
|
$
|
(13,912)
|
|
$
|
(6,672)
|
|
$
|
(12,687)
|
|
$
|
(22,303)
|
|
$
|
(25,391)
|
|
$
|
(20,942)
|
|
$
|
(22,272)
|
|
|
Net loss per share, basic and diluted
|
|
$
|
(0.21)
|
|
$
|
(0.16)
|
|
$
|
(0.08)
|
|
$
|
(0.14)
|
|
$
|
(0.25)
|
|
$
|
(0.29)
|
|
$
|
(0.24)
|
|
$
|
(0.25)
|
|
|
Weighted average shares used in computing net loss per share, basic and diluted
|
|
|
88,043
|
|
|
88,137
|
|
|
88,506
|
|
|
89,038
|
|
|
87,526
|
|
|
87,532
|
|
|
87,793
|
|
|
87,793
|
|
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of our disclosure controls and procedures, as such term is defined under Rule 13a‑15(e) promulgated under the Exchange Act. Based on this evaluation, our principal executive officer and our principal financial officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this Annual Report.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a‑15(f). Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2015.
The effectiveness of our internal control over financial reporting as of December 31, 2015 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in its attestation report which is set forth below in this Annual Report on Form 10‑K.
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Rigel Pharmaceuticals, Inc.
We have audited Rigel Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). Rigel Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Rigel Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of Rigel Pharmaceuticals, Inc. as of December 31, 2015 and 2014, and the related statements of operations, comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2015 of Rigel Pharmaceuticals, Inc. and our report dated March 8, 2016 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Redwood City, California
March 8, 2016
Changes in Internal Controls over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the fourth quarter of 2015 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations on Effectiveness of Controls and Procedures
In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Item 9B. Other Information
None.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Information regarding our directors, executive officers and corporate governance is incorporated by reference to the information set forth under the captions “Election of Directors” and “Management—Executive Officers” in our Proxy Statement for the 2016 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2015. Such information is incorporated herein by reference.
In 2003, we adopted a code of ethics, the Rigel Pharmaceuticals, Inc. Code of Conduct, which applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. Our Code of Conduct is on our website at http://media.corporate‑ir.net/media_files/IROL/12/120936/corpgov/codeofconduct.pdf. If we make any amendments to the code or grant any waiver from a provision of the code applicable to any executive officer or director, we intend to satisfy the disclosure requirement under Item 5.05 of Form 8‑K by disclosing the nature of the amendment or waiver on our website at the address and the location specified above.
Information regarding compliance with Section 16(a) of the Exchange Act is incorporated by reference to the information set forth under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in our Proxy Statement for the 2016 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2015. Such information is incorporated herein by reference.
Item 11. Executive Compensation
Information regarding executive and director compensation is incorporated by reference to the information set forth under the captions “Compensation Discussion and Analysis,” “Executive Compensation” and “Director Compensation” in our Proxy Statement for the 2016 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2015. Such information is incorporated herein by reference.
Information regarding Compensation Committee interlocks and insider participation is incorporated by reference to the information set forth under the caption “Compensation Committee Interlocks and Insider Participation” in our Proxy Statement for the 2016 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2015. Such information is incorporated herein by reference.
Information regarding our Compensation Committee’s review and discussion of our Compensation Discussion and Analysis is incorporated by reference to the information set forth under the caption “Compensation Committee Report” in our Proxy Statement for the 2016 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2015. Such information is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Information regarding security ownership of certain beneficial owners and management and securities authorized for issuance under our equity compensation plans is incorporated by reference to the information set forth under the caption “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” and “Equity Compensation Plan Information” in our Proxy Statement for the 2016 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2015. Such information is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Information regarding certain relationships and related transactions and director independence is incorporated by reference to the information set forth under the captions “Transactions with Related Persons” and “Information Regarding the Board of Directors and Corporate Governance” in our Proxy Statement for the 2016 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2015. Such information is incorporated herein by reference.
Item 14. Principal Accounting Fees and Services
Information regarding principal accounting fees and services is incorporated by reference to the information set forth under the caption “Ratification of Selection of Independent Registered Public Accounting Firm” in our Proxy Statement for the 2016 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2015. Such information is incorporated herein by reference.
PART IV
Item 15. Exhibits, Financial Statement Schedules
|
(a)
| |
The following documents are being filed as part of this Annual Report on Form 10‑K: |
|
1.
| |
Financial Statements—Index to Financial Statements in Item 8 of this Annual Report on Form 10‑K including selected quarterly financial data for the last two years in Note 13. |
|
2.
| |
Financial Statement Schedules—None—As all required disclosures have been made in the footnotes to the financial statements. |
|
3.
| |
See Exhibit Index at the end of this Annual Report, which is incorporated herein by reference. The Exhibits listed in the accompanying Exhibit Index are filed as part of this report. |
SIGNATURES
Pursuant to the requirements of the Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Annual Report on Form 10‑K to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of South San Francisco, State of California, on March 8, 2016.
|
|
|
|
|
|
Rigel Pharmaceuticals, Inc.
|
|
|
|
|
|
By:
|
/s/ Raul R. Rodriguez
|
|
|
|
Raul R. Rodriguez
Chief Executive Officer
|
|
|
|
|
|
|
By:
|
/s/ Ryan D. Maynard
|
|
|
|
Ryan D. Maynard
Executive Vice President and Chief Financial Officer
|
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Raul R. Rodriguez and Ryan D. Maynard, and each of them, as his true and lawful attorneys‑in‑fact and agents, with full power of substitution and resubstitution, for him and in his name, place, and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10‑K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys‑in‑fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming that all said attorneys‑in‑fact and agents, or any of them or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10‑K has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
/s/ Raul R. Rodriguez
|
|
Chief Executive Officer and Director
|
|
March 8, 2016
|
|
Raul R. Rodriguez
|
|
(Principal Executive Officer)
|
|
|
|
|
|
|
|
|
|
/s/ Ryan D. Maynard
|
|
Executive Vice President and Chief Financial Officer
|
|
March 8, 2016
|
|
Ryan D. Maynard
|
|
(Principal Finance and Accounting Officer)
|
|
|
|
|
|
|
|
|
|
/s/ Donald G. Payan
|
|
Executive Vice President, President of
|
|
March 8, 2016
|
|
Donald G. Payan
|
|
Discovery and Research, and Director
|
|
|
|
|
|
|
|
|
|
/s/ Gary A. Lyons
|
|
Chairman of the Board
|
|
March 8, 2016
|
|
Gary A. Lyons
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Bradford S. Goodwin
|
|
Director
|
|
March 8, 2016
|
|
Bradford S. Goodwin
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Keith A. Katkin
|
|
Director
|
|
March 8, 2016
|
|
Keith A. Katkin
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Walter H. Moos
|
|
Director
|
|
March 8, 2016
|
|
Walter H. Moos
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Peter S. Ringrose
|
|
Director
|
|
March 8, 2016
|
|
Peter S. Ringrose
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Stephen A. Sherwin
|
|
Director
|
|
March 8, 2016
|
|
Stephen A. Sherwin
|
|
|
|
|
EXHIBIT INDEX
|
|
|
| 3.1
|
Amended and Restated Certificate of Incorporation (filed as an exhibit to Rigel’s Current Report on Form 8‑ K (No. 000‑29889) dated May 29, 2012, and incorporated herein by reference).
|
|
|
|
| 3.2
|
Amended and Restated Bylaws (filed as an exhibit to Rigel’s Current Report on Form 8‑K (No. 000‑ 29889), dated February 2, 2007, and incorporated herein by reference).
|
|
|
|
| 4.1
|
Form of warrant to purchase shares of common stock (filed as an exhibit to Rigel’s Registration Statement on Form S‑1 (No. 333‑45864), as amended, and incorporated herein by reference).
|
|
|
|
| 4.2
|
Specimen Common Stock Certificate (filed as an exhibit to Rigel’s Current Report on Form 8‑K (No. 000‑29889) dated June 24, 2003, and incorporated herein by reference).
|
|
|
|
| 4.3
|
Warrant issued to HCP BTC, LLC for the purchase of shares of common stock (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended March 31, 2009 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.1+
|
Form of Stock Option Agreement pursuant to 2000 Equity Incentive Plan (filed as an exhibit to Rigel’s Registration Statement on Form S‑1 (No. 333‑45864), as amended, and incorporated herein by reference).
|
|
|
|
| 10.2
|
Collaboration Agreement between Rigel and Janssen Pharmaceutical N.V., dated December 4, 1998 (filed as an exhibit to Rigel’s Registration Statement on Form S‑1 (No. 333‑45864), as amended, and incorporated herein by reference).
|
|
|
|
| 10.3
|
Collaborative Research and License Agreement between Rigel and Pfizer Inc., dated January 31, 1999 (filed as an exhibit to Rigel’s Registration Statement on Form S‑1 (No. 333‑45864), as amended, and incorporated herein by reference).
|
|
|
|
| 10.4
|
Collaboration Agreement between Rigel and Novartis Pharma AG, dated May 26, 1999 (filed as an exhibit to Rigel’s Registration Statement on Form S‑1 (No. 333‑45864), as amended, and incorporated herein by reference).
|
|
|
|
| 10.5
|
Build‑to‑Suit Lease between Rigel and Slough BTC, LLC, dated May 16, 2001 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended June 30, 2001 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.6*
|
Amendment to Build‑to‑Suit Lease between Rigel and Slough BTC, LLC, dated October 18, 2002 (filed as an exhibit to Rigel’s Annual Report on Form 10‑K, as amended, for the fiscal year ended December 31, 2002 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
| 10.7
|
Amendment No. Two to Build‑to‑Suit Lease between Rigel and Slough BTC, LLC, dated January 31, 2005 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended September 30, 2009 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
| 10.8
|
Amendment No. Three to Build‑to‑Suit Lease between Rigel and Slough BTC, LLC, dated January 31, 2005 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended September 30, 2009 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
| 10.9
|
Amendment No. Four to Build‑to‑Suit Lease between Rigel and HCP BTC, LLC, dated February 1, 2009 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended March 31, 2009 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
| 10.10
|
First Amendment to the Collaboration Agreement between Rigel and Novartis Pharma AG, dated May 18, 2001 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended June 30, 2001 (No. 000‑ 29889) and incorporated herein by reference).
|
|
|
|
|
10.11*
|
Second Amendment to the Collaboration Agreement between Rigel and Novartis Pharma AG, dated July 6, 2001 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended September 30, 2001 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
| 10.12
|
First Amendment to the Collaboration Agreement by and between Rigel and Janssen Pharmaceutical N.V., dated June 30, 2000 (filed as an exhibit to Rigel’s Annual Report on Form 10‑K for the fiscal year ended December 31, 2001 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
| 10.13
|
Second Amendment to the Collaboration Agreement by and between Rigel and Janssen Pharmaceutical N.V., dated December 4, 2001 (filed as an exhibit to Rigel’s Annual Report on Form 10‑K for the fiscal year ended December 31, 2001 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.14*
|
Collaboration Agreement between Rigel and Daiichi Pharmaceutical Co., Ltd., dated August 1, 2002 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended September 30, 2002 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.15+
|
Employment Agreement between Rigel and Elliott B. Grossbard, dated as of March 18, 2002 (filed as an exhibit to Rigel’s Annual Report on Form 10‑K, as amended, for the fiscal year ended December 31, 2002 (No. 000‑ 29889) and incorporated herein by reference).
|
|
|
|
|
10.16*
|
Collaborative Research and License Agreement by and between Rigel and Pfizer Inc., dated January 18, 2005 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended March 31, 2005 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.17+
|
Form of Indemnity Agreement (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended March 31, 2007 (No. 000‑29889), as amended, and incorporated herein by reference).
|
|
|
|
|
10.18+
|
2000 Equity Incentive Plan, as amended (filed as an exhibit to Rigel’s Registration Statement on Form S‑8 (No. 333‑189523) filed on June 21, 2013 and incorporated herein by reference).
|
|
|
|
|
10.19+
|
2000 Non‑Employee Directors’ Stock Option Plan, as amended (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended June 30, 2015 (No. 000‑29889) filed on August 4, 2015 and incorporated herein by reference).
|
|
|
|
|
10.20+
|
Amended and Restated Employment Agreement between Rigel and Donald G. Payan, effective January 1, 2011 (filed as an exhibit to Rigel’s Annual Report on Form 10‑K for the fiscal year ended December 31, 2010 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.21+
|
Amended and Restated Change of Control Severance Plan (filed as an exhibit to Rigel’s Annual Report on Form 10‑K for the fiscal year ended December 31, 2010 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.22+
|
2000 Employee Stock Purchase Plan, as amended (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended March 31, 2010 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.23*
|
License and Collaboration Agreement between Rigel and AstraZeneca AB, dated February 15, 2010 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended March 31, 2010 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.24+
|
2011 Equity Incentive Plan, as amended (filed as an exhibit to Rigel’s Registration Statement on Form S‑8 (No. 333‑189523) filed on June 21, 2013 and incorporated herein by reference).
|
|
|
|
|
10.25*
|
Termination Agreement between Rigel and Pfizer, Inc., dated May 2, 2011 (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended June 30, 2011 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.26+
|
Form of Stock Option Agreement pursuant to 2011 Equity Incentive Plan (filed as an exhibit to Rigel’s Quarterly Report on Form 10‑Q for the quarter ended September 30, 2011 (No. 000‑29889) and incorporated herein by reference).
|
|
|
|
|
10.27+
|
2012 Cash Incentive Plan (filed as an exhibit to Rigel’s Current Report on Form 8‑K (No. 000‑29889) filed on February 8, 2012, and incorporated herein by reference).
|
|
|
|
|
10.28+
|
2013 Cash Incentive Plan (filed as an exhibit to Rigel’s Current Report on Form 8‑K (No. 000‑29889) filed on February 14, 2013, and incorporated herein by reference).
|
|
|
|
|
10.29+
|
2014 Cash Incentive Plan (filed as an exhibit to Rigel’s Current Report on Form 8‑K (No. 000‑29889) filed on May 20, 2014, and incorporated herein by reference).
|
|
|
|
|
10.30+
|
2015 Cash Incentive Plan (filed as an exhibit to Rigel’s Current Report on Form 8‑K (No. 000‑29889) filed on January 30, 2015, and incorporated herein by reference).
|
|
|
|
| 10.31
|
Controlled Equity OfferingSM Sales Agreement, dated August 18, 2015, by and between Rigel Pharmaceuticals, Inc. and Cantor Fitzgerald & Co. (filed as an exhibit to Rigel’s Current Report on Form 8‑K (No. 000‑29889) filed on August 18, 2015, and incorporated herein by reference).
|
|
|
|
|
23.1#
|
Consent of Independent Registered Public Accounting Firm.
|
|
|
|
|
24.1#
|
Power of Attorney (included on signature page).
|
|
|
|
|
31.1#
|
Certification required by Rule 13a‑14(a) or Rule 15d‑14(a).
|
|
|
|
|
31.2#
|
Certification required by Rule 13a‑14(a) or Rule 15d‑14(a).
|
|
|
|
|
32.1•
|
Certification required by Rule 13a‑14(b) or Rule 15d‑14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. 1350).
|
|
|
|
|
101.INS#
|
XBRL Instance Document
|
|
|
|
|
101.SCH#
|
XBRL Taxonomy Extension Schema Document
|
|
|
|
|
101.CAL#
|
XBRL Taxonomy Extension Calculation Linkbase Document
|
|
|
|
|
101.LAB#
|
XBRL Taxonomy Extension Labels Linkbase Document
|
|
|
|
|
101.PRE#
|
XBRL Taxonomy Extension Presentation Linkbase Document
|
|
|
|
|
101.DEF#
|
XBRL Taxonomy Extension Definition Linkbase Document
|
+Management contract or compensatory plan.
*Confidential treatment requested as to specific portions, which portions are omitted and filed separately with the Securities and Exchange Commission.
#Filed herewith.
•The certification attached as Exhibit 32.1 accompanies the Annual Report on Form 10‑K pursuant to Section 906 of the Sarbanes‑Oxley Act of 2002 and shall not be deemed “filed” by the Company for purposes of Section 18 of the Securities Exchange Act of 1934, as amended.